
线纹海马肠道菌群结构与功能对迟钝爱德华氏菌(Edwardsiella tarda)感染的响应特征研究
|
张乐乐(1998—), 女, 山东省聊城市人, 硕士研究生, 主要从事海洋动物病害研究。email: |
收稿日期: 2021-06-11
修回日期: 2021-07-22
网络出版日期: 2021-07-26
基金资助
山东省高等学校优秀青年创新团队科技计划项目(2020KJF007)
烟台市科技创新发展计划项目(2020LJRC120)
烟台市科技创新发展计划项目(2019CXJJ040)
山东省高校科技计划项目(J18KA146)
LAMB联合资助开放基金课题(LMB20200103)
威海市科技发展计划项目(2017GNS10)
Dysbiosis of both structure and function of intestinal microbiota in lined seahorses (Hippocampus erectus) as response to Edwardsiella tarda infection
Received date: 2021-06-11
Revised date: 2021-07-22
Online published: 2021-07-26
Supported by
Program for Outstanding Youth of Colleges and Universities(2020KJF007)
Yantai Foundation for Development of Science and Technology(2020LJRC120)
Yantai Foundation for Development of Science and Technology(2019CXJJ040)
Shandong Province Science and Technology Research Program for Colleges and Universities(J18KA146)
LMM
LMB, LMM and LAMB Co-funded Open Funds of the South China Sea Institute of the Chinese Academy of Sciences(LMB20200103)
Weihai Foundation for Development of Science and Technology(2017GNS10)
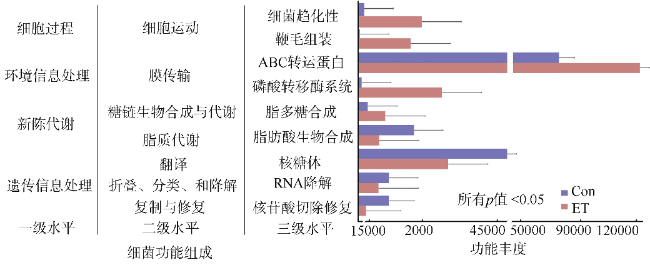
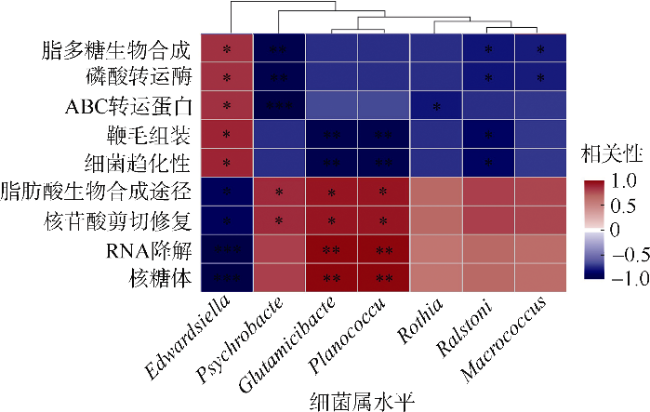
细菌性肠炎对海马养殖业影响巨大, 但病原对海马肠道菌群的具体影响尚不清楚。文章利用已分离的病原细菌 Edwardsiella tarda YT1和海马细菌性肠炎模型, 结合16S rDNA高通量测序技术, 探究病原细菌侵染对海马肠道菌群的影响。结果发现, E. tarda侵染改变了海马肠道菌群的结构组成、多样性和丰度, 并显著降低了其多样性(p<0.05); 显著增加了海马肠道变形菌门(Proteobacteria)的相对丰度(p<0.05), 减少了放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)的相对丰度(p<0.05); 导致致病菌爱德华氏菌属(Edwardsiella)在属水平的相对丰度极显著增加(p<0.01), 而肠道固有菌群嗜冷杆菌属(Psychrobacter)和罗氏菌属(Rothia)极显著减少(p<0.01), 以及球菌属(Macrococcus)与动球菌属(Planococcus)显著减少(p<0.05)。研究结果表明, E. tarda能通过改变海马肠道固有优势菌群的相对丰度导致菌群失调。菌群功能变化及其相关性分析表明, E. tarda可能通过显著提高细菌趋化性、鞭毛组装、ABC转运蛋白、磷酸转运酶系统以及脂多糖生物合成途径的活性(p<0.05), 抑制肠道核心菌群如嗜冷杆菌属、动球菌属和谷氨酸杆菌属的丰度及其核糖体、RNA降解、核苷酸剪切修复与脂肪酸生物合成途径的活性(p<0.05), 导致肠道菌群功能失调, 并诱发肠炎。
张乐乐 , 邹强 , 田雅楠 , 吕春晖 , 郑诗怡 , 姜广峻 , 高龙坤 , 侯玉平 , 王凯 . 线纹海马肠道菌群结构与功能对迟钝爱德华氏菌(Edwardsiella tarda)感染的响应特征研究[J]. 热带海洋学报, 2022 , 41(2) : 177 -188 . DOI: 10.11978/2021074
Bacterial enteritis can cause severe damage to seahorse aquaculture, while little is known about the effects of bacterial pathogen infection on intestinal microbiota of seahorses. In the present study, both bacterial pathogen (Edwardsiella tarda YT1) and seahorse research model of bacterial enteritis previously reported by us were employed to explore the role of bacterial pathogen in intestinal microbiota by high-throughput full-length 16S rRNA gene sequencing. The results showed that E. tarda infection significantly altered the composition and abundance, and significantly decreased diversity of intestinal microbiota of lined seahorses (p<0.05); significantly increased the relative abundance of Proteobacteria (p<0.05), and decreased the abundance of Actinobacteria, Firmicutes, and Bacteroidetes (p<0.05); significantly increased the relative abundance of pathogenic Edwardsiella (p<0.01), while decreasing that of Psychrobacter, Rothia, Macrococcus, and Planococcus (p<0.05) at genera level. It indicates that E. tarda infection can reduce the diversity and relative abundance of intestinal autochthonous microbiota and result in dysbiosis in lined seahorses. Based on the results of bacterial function and correlation, increasing relatively abundance of E. tarda may significantly upregulate its activities of bacterial chemotaxis, flagella assembly, ABC transporter, phosphotransferase system, and lipopolysaccharide biosynthesis pathways (p<0.05), cause decrease of the relative abundance of the core intestinal microbiota of Psychrobacter, Planococcus and Glutamicibacter and suppression of their functional activities of ribosome, RNA degradation, nucleotide excision repair, and fatty acid biosynthesis pathways (p<0.05), induce dysbiosis of intestinal microbiota, and finally result in enteritis. These results may be helpful for further revealing pathogenic mechanism of E. tarda-induced enteritis in seahorses by more detailed metagenomic and metabolomics analysis.
Key words: seahorse; intestinal microbiota; enteritis; dysbiosis; Edwardsiella tarda
表1 样本信息统计表Tab. 1 Statistics of sequencing data from each sample |
| 样本名称 | 序列数/bp | 碱基数/bp | 平均长度/bp | 最短序列长度/bp | 最长序列长度/bp |
|---|---|---|---|---|---|
| Con_1 | 40963 | 17215829 | 420.28 | 211 | 478 |
| Con_2 | 89292 | 37497350 | 419.94 | 208 | 527 |
| Con_3 | 53207 | 22541120 | 423.65 | 211 | 524 |
| ET_1 | 31899 | 13629173 | 427.26 | 211 | 432 |
| ET_2 | 48727 | 20886268 | 428.64 | 211 | 431 |
| ET_3 | 36722 | 15739755 | 428.62 | 319 | 430 |
注: Con为PBS注射对照组; ET为Edwardsiella tarda YT1胁迫组 |
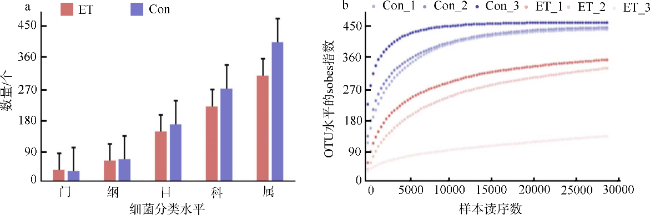
图1 Edwardsiella tarda YT1侵染前后海马肠道菌群16S rDNA测序分析a. OTU统计; b. 基于OTUs绘制的稀释曲线。Con为PBS注射对照组; ET为E. tarda YT1胁迫组 Fig. 1 Basic analysis of 16S rDNA sequencing of intestinal microbiota during Edwardsiella tarda YT1 infection of Hippocampus erectus. (a) OTU statistics; (b) Rarefaction curves of OTUs of intestinal microbiota of sampled seahorses |
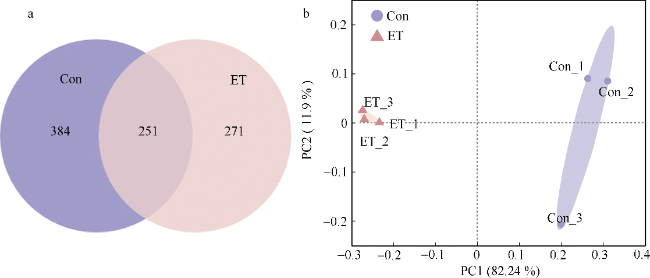
图2 Edwardsiella tarda YT1侵染对海马肠道菌群组成结构的影响a. OUT水平Venn图; b. 肠道菌群的PCoA加权分析。Con为PBS注射对照组; ET为E. tarda YT1胁迫组 Fig. 2 Effect of Edwardsiella tarda YT1 infection on composition and structure of intestinal microbiota of lined seahorse. (a) The diagram of Venn at OUT level; (b) PCoA of weighted UniFrac distance matrices of intestinal microbiota |
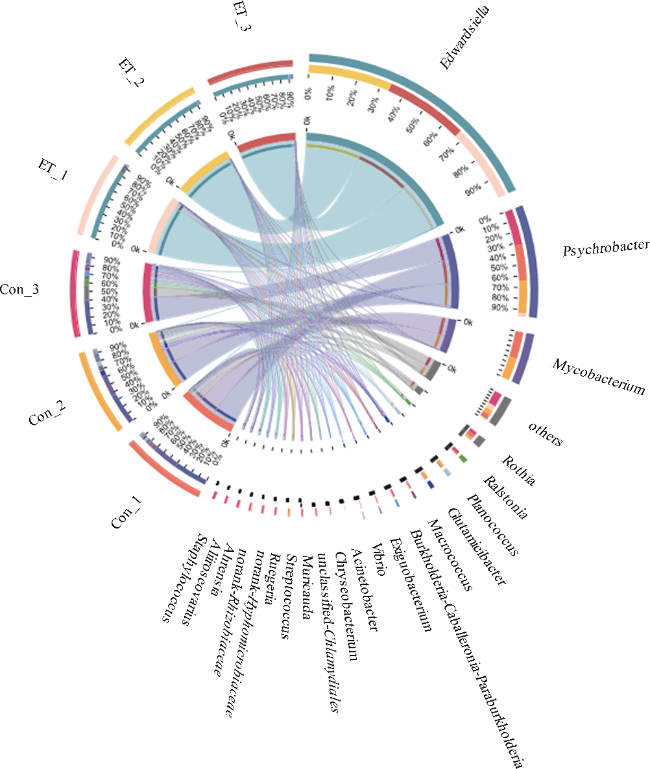
图4 Edwardsiella tarda YT1侵染对海马肠道菌群(属水平)相对丰度的影响内、外圈分别显示肠道菌群在属水平的相对丰度与名称。Con为PBS注射对照组; ET为E. tarda YT1胁迫组 Fig. 4 Comparison of intestinal microbiota between healthy and diseased seahorses at genus level (relative abundance > 1%) by circular representation. The inner and outer circular diagrams show the relative abundance of intestinal microbiota at genus level and name, respectively |
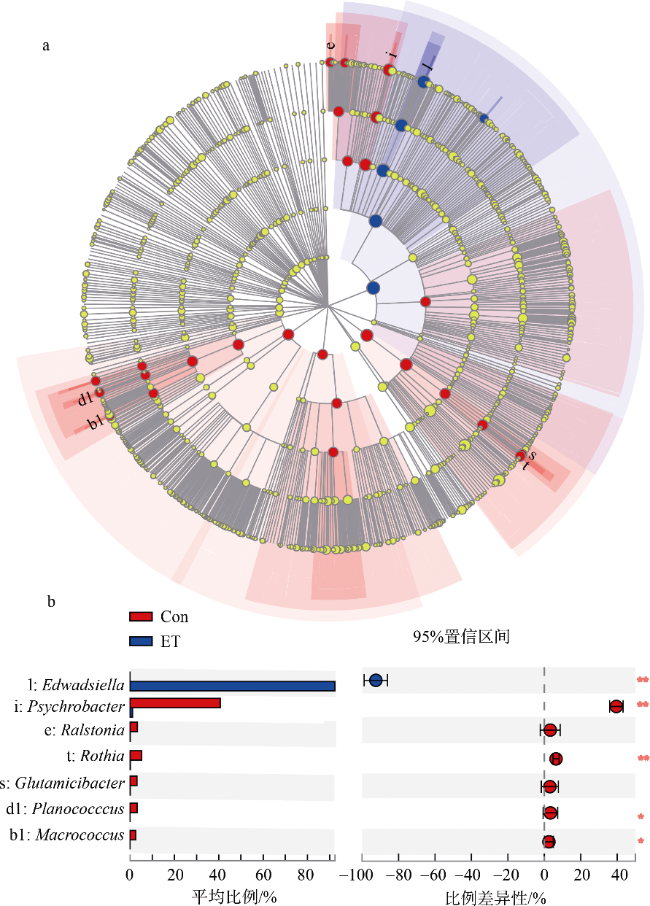
图5 Edwardsiella tarda YT1侵染对肠道中优势菌群相对丰度的影响a. 基于线性判别分析效应(LEfSe)绘制的海马肠道微生物群落的物种群落结构图, 图中字母指代菌属见图b。不同颜色节点表示在对应组别中显著富集, 且对组间差异存在显著影响的微生物类群; 淡黄色节点表示在不同分组中均无显著差异或对组间差异无显著影响的微生物类群。节点大小显示了物种的丰度; b. 差异明显的属的定量分析。*表示p<0.05, **表示p<0.01。Con为PBS注射对照组; ET为E. tarda YT1胁迫组 Fig. 5 Comparison of dominant intestinal microbiota between healthy and diseased seahorses. (a) Species community structure diagram of gut microbiomes of the seahorse by linear discriminant analysis effect size (LEfSe); (b) Quantitative analysis of genus (* p<0.05, ** p<0.01). The node’s size displays the abundance of the species |
图6 Edwardsiella tarda YT1侵染对海马肠道菌群功能的影响Con为PBS注射对照组; ET为E. tarda YT1胁迫组 Fig. 6 Functional differences of intestinal microbiota during Edwardsiella tarda YT1-induced enteritis in lined seahorses |
图7 病原侵染过程中海马肠道优势菌属与功能的相关性分析红色代表优势菌属与功能呈正相关, 蓝色代表负相关。*表示p<0.05, **表示p<0.01, ***表示p<0.001。图上方的聚类分析表示肠道优势菌属共分为两大支, 其中Edwardsiella为一支, 其他优势属为一支 Fig. 7 Correlation between relative abundance of dominant genera and functions of intestinal microbiota during Edwardsiella tarda YT1-induced enteritis in lined seahorses (* p<0.05, ** p<0.01, *** p<0.001) |
表2 迟钝爱德华氏菌的侵染对海马肠道菌群α多样性的影响Tab. 2 Effects of Edwardsiella tarda YT1 infection on α diversity of intestinal microbiota of lined seahorses |
| 名 称 | Con | ET | p值 |
|---|---|---|---|
| Shannon指数 | 2.59±0.65a | 0.89±0.53b | 0.02 |
| Simpson指数 | 0.19±0.06b | 0.68±0.28a | 0.04 |
| ACE指数 | 449.82±31.96 | 362.46±232.13 | 0.55 |
| Chao指数 | 439.46±41.06 | 326.82±245.5 | 0.47 |
| 物种覆盖度 | 0.99 | 0.99 | 0.39 |
注: Con为PBS注射对照组; ET为E. tarda YT1胁迫组。变量值表示为“平均值±标准误”, 同列上标字母相同表示数据间差异不显著(p>0.05) |
| [1] |
邓钢, 吕军仪, 林强, 2005. 大海马育苗池水华发生期间细菌动态及相关理化参数[J]. 中国水产科学, 12(4): 477-482.
|
| [2] |
李焕宇, 付婷婷, 张云, 等, 2017. 5种方法提取真菌基因组DNA作为PCR模板效果的比较[J]. 中国农学通报, 33(16): 28-35.
|
| [3] |
刘国信, 茹贺军, 2006. 海马的人工养殖技术[J]. 水产科技情报, 33(6): 254-256.
|
| [4] |
吕军仪, 吴金英, 杨大伟, 等, 2001. 大海马在人工养殖条件下的生长速率[J]. 中国水产科学, 8(1): 59-63.
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}