
基于RNA-Seq高通量测序技术的法螺差异表达基因分析
|
刘文广(1978—), 山东省聊城市人, 副研究员, 主要从事贝类育种及环境适应性研究。email: |
Copy editor: 姚衍桃
收稿日期: 2022-09-20
修回日期: 2022-10-17
网络出版日期: 2022-10-13
基金资助
国家自然科学基金项目(42176129)
广东省自然科学基金项目(2022A1515010779)
Analysis of differential expressed genes from Charonia tritonis based on transcriptome sequencing
Copy editor: YAO Yantao
Received date: 2022-09-20
Revised date: 2022-10-17
Online published: 2022-10-13
Supported by
National Natural Science Foundation of China(42176129)
Natural Science Foundation of Guangdong Province(2022A1515010779)
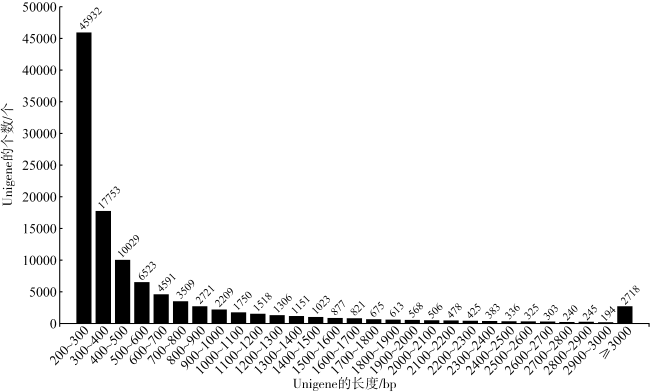
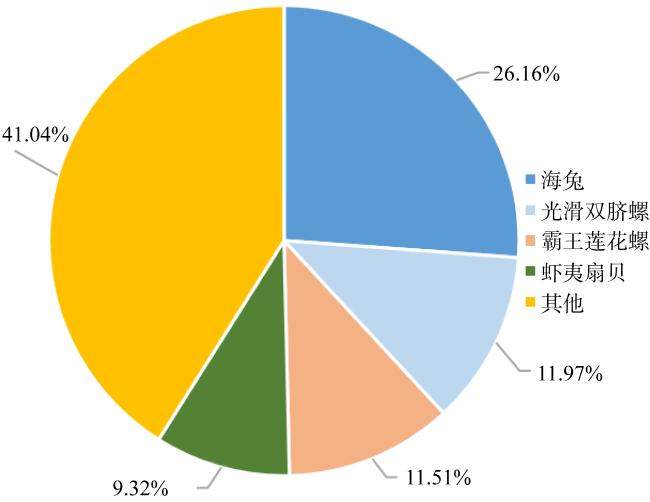
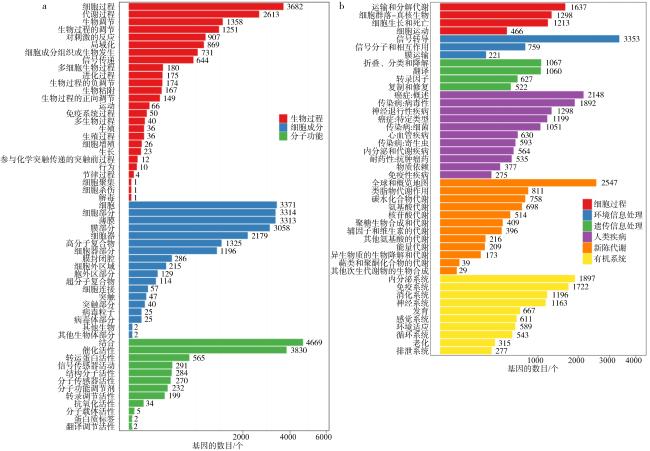
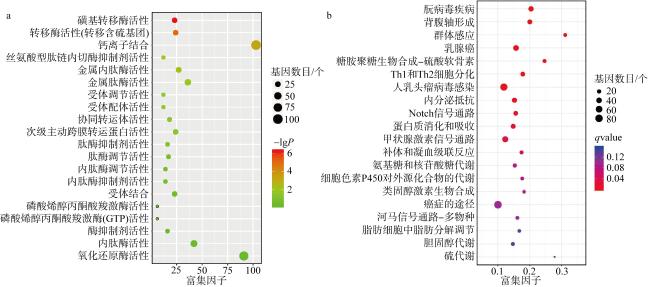
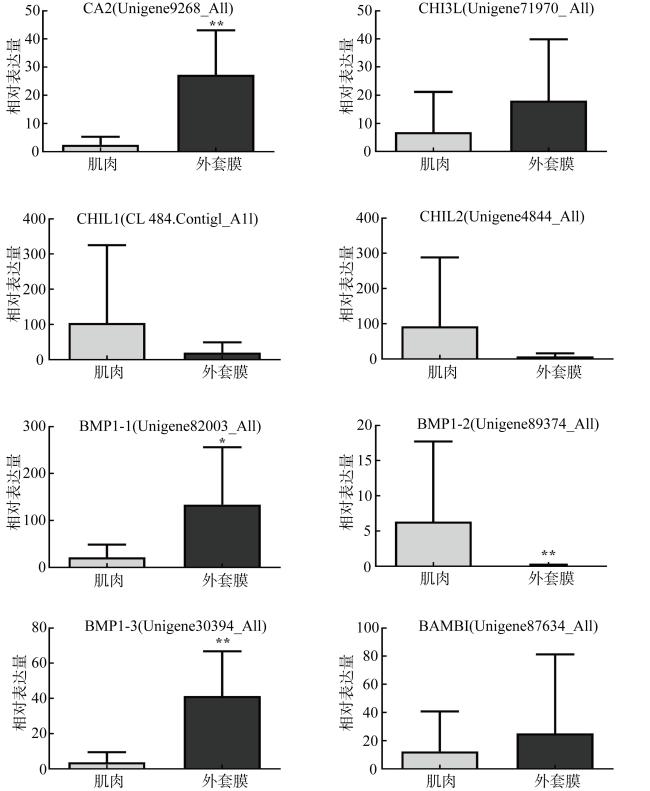
法螺是珊瑚礁“杀手”长棘海星为数不多的自然捕食者之一, 是珊瑚礁生态系统的重要保卫者。但是, 由于海洋环境变化、人类的过度捕捞、珊瑚礁栖息地被破坏等因素, 法螺的自然资源遭受严重破坏, 已长期处于濒临灭绝的状态。尽快开展法螺基础研究迫在眉睫, 但是该物种的基因组资源有限, 阻碍了如生物矿化等分子机制的深入研究。本研究借助于Illumina HiSeqTM2000测序平台完成了法螺两种组织(肌肉与外套膜)的高通量测序工作。测序所得原始序列经筛选、组装之后比对到NR、NT、Swissprot、KEGG、GO和InterPro数据库中获得注释结果, 并进行差异基因聚类分析。结果表明, 测序共获得109722个Unigene, 其中有46239个Unigene得到功能注释, 共获得7994个差异表达基因, 其中在法螺外套膜组织中有 3196个上调, 4798个下调。对差异表达基因(differentially expressed genes, DEG)的GO注释和KEGG富集分析显示, 高表达的DEG显著富集在典型的生物矿化相关通路中, 如糖胺聚糖生物合成硫酸软骨素通路(glycosaminoglycan biosynthesis chondroitin sulfase dermatan sulfate)、磺基转移酶活性(sulfotransferase activity)、钙离子结合(calcium ion binding)等。通过实时定量荧光聚合酶链反应(RT-qPCR)对8个与矿化相关的DEG基因的表达情况进行验证表明, 其表达趋势与转录组测序结果一致。本研究中所获得丰富的转录本和全面的转录组信息, 增加了法螺基因信息的同时, 还为法螺矿化机制的解析提供了一定的数据支持。
刘文广 , 张格格 , 岑希彤 , 何毛贤 . 基于RNA-Seq高通量测序技术的法螺差异表达基因分析[J]. 热带海洋学报, 2023 , 42(4) : 146 -154 . DOI: 10.11978/2022197
The giant triton snail (Charonia tritonis), an endangered gastropod species of ecological and economic importance, is widely distributed in coral reef ecosystems of the Indo-West Pacific region and the tropical waters of the South China Sea. Research on molecular mechanisms is limited due to the lack of complete genomic data for this species. In the present work, transcriptome sequencing of foot muscle and mantle in C. tritonis were obtained by Illumina HiSeq sequencing platform, from which 7994 (3196 upregulated and 4798 downregulated) differentially expressed genes (DEGs) containing biomineralization sequences were identified. The top 20 GO terms in the molecular function category were considered to be related to biomineralization. In KEGG classifications, DEGs are primarily enriched in some pathways that may be involved in biomineralization. The results of qPCR showed that three of the eight genes examined are significantly up-regulated in the mantle. Our results improve the understanding of biomineralization in C. tritonis and provide fundamental transcriptome information to study other molecular mechanisms, such as reproduction.
Key words: Charonia tritonis; transcriptome; DEG
表1 转录组数据组装结果统计Tab. 1 Assembly result of transcriptome data |
| 样本名称 | 总数/个 | 总长度/bp | 平均长度/bp | N50长度/bp | N70长度/bp | GC碱基对数量/% |
|---|---|---|---|---|---|---|
| 法螺肌肉 | 87443 | 50845674 | 581 | 799 | 426 | 43.26 |
| 法螺外套膜 | 81100 | 48450379 | 597 | 843 | 442 | 43.78 |
| All-Unigene | 109722 | 70289861 | 640 | 995 | 485 | 43.43 |
表2 基因注释数量统计Tab. 2 Number of Unigene annotated in NR, NT, Swissprot, KEGG, GO and InterPro database |
| 总数 | NR | NT | Swissport | KEGG | KOG | InterPro | GO | Intersection | Overall | |
|---|---|---|---|---|---|---|---|---|---|---|
| 基因个数 | 109722 | 26050 | 27059 | 16944 | 18705 | 15319 | 17617 | 9788 | 2712 | 46239 |
| 注释比例 | 100% | 23.74% | 24.66% | 15.44% | 17.05% | 13.96% | 16.06% | 8.92% | 2.47% | 42.14% |
注: Intersection指的是7个数据库中同时存在的Unigene的数量和比例; Overall为7个数据库中所有数据库注释上的Unigene总数及比例 |
图5 差异表达基因的qPCR验证结果各子图标题中: CA表示carbonic anhydrase(碳酸酐酶), BMP表示bone morphogenetic protein(骨形成蛋白), BAMBI表示BMP and activin membrane bound inhibitor(BMP和激活素膜结合抑制剂), CHIL表示chitinase-like lectin(几丁质酶样凝集素), CHI3L表示chitinase-3-like protein(壳多糖酶3样蛋白); 括号中注明的是相关基因的ID Fig. 5 Relative mRNA expression profiles of biomineralization-related genes of C. tritonis |
| [1] |
姜铁民, 张勇, 黄洁, 等, 2003. 合浦珠母贝26S蛋白酶体S4, S7亚基基因片段的分离与分析[J]. 海洋科学, 27(4): 28-34.
|
| [2] |
李琛, 郝瑞娟, 王庆恒, 等, 2017. 马氏珠母贝磺基转移酶PmCHST11基因的分子特征与表达分析[J]. 基因组学与应用生物学, 36(1): 150-157.
|
| [3] |
潘肖兰, 刘惠茹, 许濛, 等, 2020. 马氏珠母贝水通道蛋白基因AQP4 cDNA 克隆和表达分析[J]. 热带海洋学报, 39(3): 66-75.
|
| [4] |
王庆恒, 郝瑞娟, 郑哲, 等, 2017. 马氏珠母贝磺基转移酶基因的克隆及功能[J]. 水产学报, 41(5): 669-677.
|
| [5] |
张格格, 2021. 法螺的遗传多样性分析及转录组测序[D]. 北京: 中国科学院大学.
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}