
热带海草泰来草沉积物真菌的群落结构、功能与分子生态网络研究
|
凌娟, 研究员, 从事微生物海洋学及资源利用研究。email: |
收稿日期: 2022-10-24
修回日期: 2022-12-09
网络出版日期: 2022-12-26
基金资助
海南省自然科学基金项目(422QN440)
国家自然科学基金项目(41676163)
国家自然科学基金项目(42276160)
国家自然科学基金项目(42206129)
广东省基础与应用基础研究基金项目(2023A1515012124)
广东省科技计划项目(2021B1212050023)
广东省科技计划项目(2020B1212060058)
Community structure, function, and molecular ecological network of fungi in the tropical seagrass Thalassia hemprichii sediment
Received date: 2022-10-24
Revised date: 2022-12-09
Online published: 2022-12-26
Supported by
Hainan Provincial Natural Science Foundation of China(422QN440)
National Natural Science Foundation of China(41676163)
National Natural Science Foundation of China(42276160)
National Natural Science Foundation of China(42206129)
Guangdong Basic and Applied Basic Research Foundation(2023A1515012124)
Science and Technology Planning Project of Guangdong Province of China(2021B1212050023)
Science and Technology Planning Project of Guangdong Province of China(2020B1212060058)
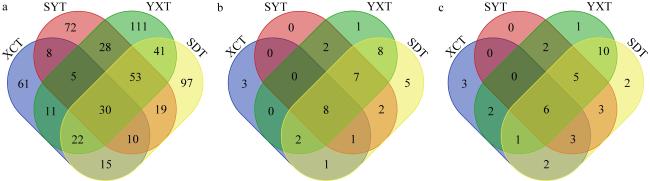
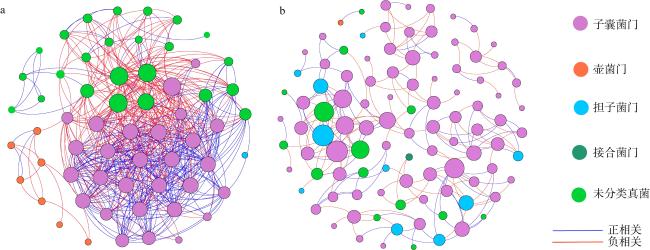
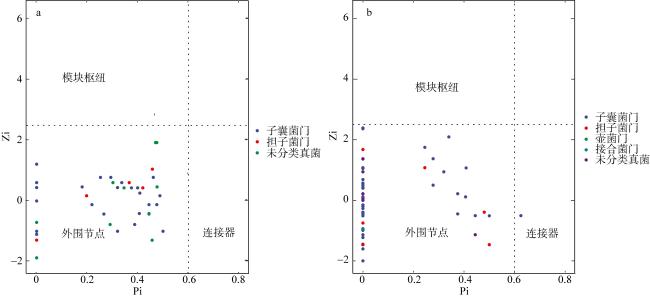
真菌是海草床生态系统的重要组成部分, 它们在维持海草健康和生态系统碳氮等营养元素循环方面发挥着重要作用。为了阐明海草床生态系统真菌的群落结构及功能, 文章以热带海草泰来草(Thalassia hemprichii)沉积物为主要研究对象, 通过Illumina Miseq高通量测序技术分析海南岛和西沙群岛两个研究区域泰来草沉积物真菌的群落结构与物种多样性, 并利用FUNGuild数据库对真菌营养类型进行预测和功能注释。研究结果表明, 子囊菌门(Ascomycota)(相对丰度24.30%~76.20%)和担子菌门(Basidiomycota)(相对丰度4.98%~52.24%)为两个研究区域的共同优势种群, 但是子囊菌门真菌的相对丰度在两个研究区域间存在显著性差异(p<0.05), 两个区域内共有的海草沉积物真菌分类操作单元(operational taxonomic units, OTUs)数量占比为5.15%, 其相对丰度为31.19%。两个研究区域泰来草沉积物真菌群落的α多样性指数中的香农指数(Shannon)和系统发育多样性(phylogenetic diversity)以及β多样性之间都存在显著性差异(p<0.05)。FUNGuild功能预测分析表明, 大部分真菌的营养类型尚未确定(72.11%~91.92%), 其中能够确定的主要营养类型为共生营养型(symbiotroph)、腐生营养型(saprotroph)和病理营养型(pathotroph), 3种营养型可以进一步分为41个生态功能群。基于随机矩阵理论(random matrix theory, RMT)构建的两个研究区域海草泰来草沉积物真菌分子生态网络结果显示, 海南岛泰来草沉积物真菌网络结构更加复杂, 其平均聚类系数更高、平均连通度更高、密度更高, 该真菌群落可能对外界环境变化更为敏感, 而西沙群岛海草沉积物真菌群落的模块化程度更高, 其中粪壳菌纲(Sordariomycetes)为分子生态网络中的关键类群。
凌娟 , 梁童茵 , 岳维忠 , 黄小芳 , 孙翠慈 , 张健 , 张煜航 , 周卫国 , 董俊德 . 热带海草泰来草沉积物真菌的群落结构、功能与分子生态网络研究[J]. 热带海洋学报, 2023 , 42(5) : 64 -75 . DOI: 10.11978/2022226
Fungi are essential components of seagrass ecosystems, and they play important roles in maintaining seagrass health and nutrient cycling in the ecosystem. To elucidate the fungal community structure and their functions in seagrass sediment, we used Illumina MiSeq high-throughput sequencing technique to investigate the fungi in sediments of tropical seagrass Thalassia hemprichii in Hainan Island and Xisha Islands, respectively. FUNGuild database was introduced to predict fungi trophic types and annotate fungi guilds. Results showed that phylum Ascomycota (relative abundance 24.30% ~ 76.20%) and Basidiomycota (relative abundance 4.98% ~ 52.24%) were the dominant phyla in the two study areas, but the relative abundance of phylum Ascomycota was significantly different between the two study areas (p < 0.05). The percentage of OTUs numbers in seagrass sediment fungi shared in the two regions was 5.15%, and their relative abundance was 31.19%. In addition, there were significant differences between the Alpha diversity index (Shannon and Phylogenetic diversity) and Beta diversity of the fungal communities of seagrass sediments in the two study areas (p < 0.05). The FUNGuild functional prediction analysis revealed that the main fungal trophic types were undetermined (relative abundance 72.11% ~ 91.92%). The trophic types of the rest fungi were Symbiotroph, Saprotroph, and Pathotroph, and these three trophic types could be further divided into 41 functional guilds. Network analysis for fungal groups based on random matrix theory (RMT) showed that the fungi network structure of seagrass T. hemprichii sediment in Hainan Island was more complex, with higher average clustering coefficients, longer average path lengths, and higher densities. These fungal communities may be more sensitive to environmental change. While the fungal communities of seagrass T. hemprichii sediment in Xisha Islands were more modulized, the fungus belonging to Class Sordariomycetes was the key taxon in the molecular ecological network. This study provides essential primary data and theoretical support for further study on the structure and function of fungi in seagrass ecosystems, microbial resource mining, and ecological applications.
表1 热带海草泰来草沉积物样品编号、采样区及其相关信息Tab. 1 The information of seagrass Thalassia hemprichii sediment |
| 序号 | 分组 | 编号 | 样品 | 采样地 | 海草种类组成 | 具体时间 | 经纬度 |
|---|---|---|---|---|---|---|---|
| 1 | 海南岛 | XCT | 泰来草 沉积物 | 海南岛陵水新村湾海草特别保护区 | 泰来草海菖蒲 | 2015年6月1日 | 18°30′N, 109°59′E |
| 2 | 海南岛 | SYT | 泰来草 沉积物 | 海南岛三亚珊瑚礁自然保护区 | 泰来草 | 2015年6月1日 | 18°12′N, 109°30′E |
| 3 | 西沙群岛 | YXT | 泰来草 沉积物 | 西沙群岛永兴岛 | 泰来草、喜盐草、 二药草 | 2015年5月28日 | 16°50′32″N, 112°20′41″E |
| 4 | 西沙群岛 | SDT | 泰来草 沉积物 | 西沙群岛石岛 | 泰来草、喜盐草 | 2015年5月28日 | 16°50'06″N, 112°22′10″E |
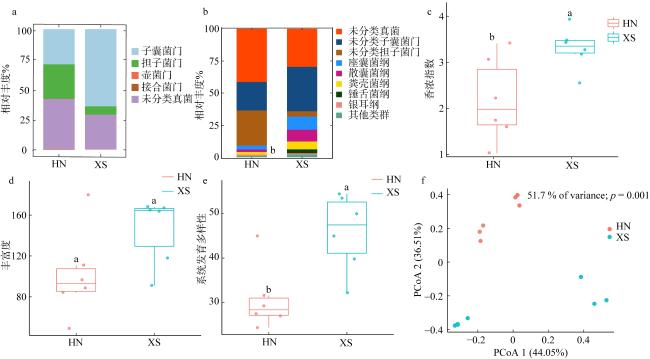
图1 海草泰来草沉积物的真菌群落组成(a~b)与α和β多样性分析(c~f)a. 门水平上的真菌群落结构组成; b. 纲水平上的真菌群落结构组成; c.香农指数; d. 丰富度; e. 系统发育多样性; f. β多样性的主坐标分析主成分PCoA分析。图c~e中相同的小写字母表示不同研究区域间无显著性差异(p>0.05)而不同小写字母代表不同研究区域间具有显著性差异(p<0.05)。HN表示海南岛; XS表示西沙群岛 Fig. 1 Fungal community composition and diversity analysis of seagrass sediments. The fungal community composition at the phylum level (a) and class level (b), respectively. The alpha diversity of fungal communities: Shannon index (c), richness (d), and phylogenetic diversity (e); principal coordinate analysis of beta diversity PCoA analysis (f) |
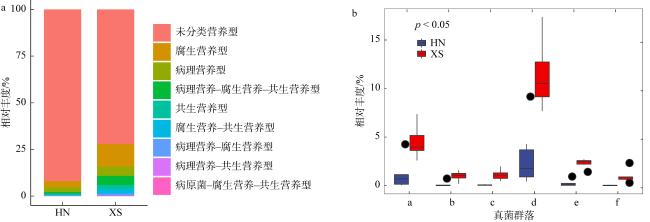
图3 海草泰来草沉积物真菌群落营养类型组成(a)及具有显著性差异的功能类群(b)分析横坐标轴的a表示植物病原菌; b表示植物病原-木腐菌; c表示真菌寄生菌-未定义腐生菌; d表示未定义腐生菌; e表示排泄物腐生菌-内生菌-凋落物腐生菌-未定义腐生菌; f表示内生菌。HN表示海南岛; XS表示西沙群岛 图b The analysis of trophic mode (a) and guilds (b) of fungal communities in seagrass sediments (a: Plant Pathogen; b: Plant Pathogen-Wood Saprotroph; c: Fungal Parasite-Undefined Saprotroph; d: Undefined Saprotroph; e: Dung Saprotroph-Endophyte-Litter Saprotroph-Undefined Saprotroph; f: Endophyte). (HN: Hainan Island; XS: Xisha Island) |
表2 海南岛和西沙群岛泰来草沉积物真菌群落分子生态网络的拓扑参数Tab. 2 Molecular ecological network topological parameters of fungal communities in the Hainan Island and Xisha Islands |
| 研究 区域 | 分子生态网络 | 随机网络 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 节点 | 连线数 | 阈值 | 平均连通度 | 平均聚集系数 | 平均路径长度 | 模块性 (模块数) | 密度 | 正相关 | 平均路径长度 | 平均 聚集系数A | 模块性 | |
| 海南岛 | 61 | 515 | 0.950 | 16.885 | 0.589 | 2.308 | 0.172(4) | 0.281 | 51.46% | 1.862±0.024 | 0.524±0.016 | 0.111±0.009 |
| 西沙群岛 | 101 | 201 | 0.890 | 3.980 | 0.257 | 4.776 | 0.740(8) | 0.040 | 57.71% | 3.433 ±0.064 | 0.040 ± 0.013 | 0.458±0.013 |
图5 海草泰来草沉积物海南岛(a)和西沙群岛(b)真菌群落结构的拓扑角色基于Zi和Pi值进行节点分类, 以识别关键种类。水平线Zi=2.5, 模块枢纽Zi> 2.5; 垂直线为Pi=0.62, 连接器为Pi>0.62, 虚线表示Zi和Pi值 Fig. 5 Topological roles of fungi in seagrass Thalassia hemprichii sediment in Hainan Island (a) and Xisha Islands (b) (Nodes are classified based on Zi and Pi values to identify the key categories. The horizontal line has a Zi value of 2.5 and the modular hub Zi is > 2.5; the vertical line is Pi value = 0.62 and the connector is Pi > 0.62, the dotted lines indicate Zi and Pi values) |
| [1] |
胡晓婧, 刘俊杰, 魏丹, 等, 2018. 东北黑土区不同纬度农田土壤真菌分子生态网络比较[J]. 应用生态学报, 29(11): 3802-3810.
|
| [2] |
黄小平, 江志坚, 张景平, 等, 2018. 全球海草的中文命名[J]. 海洋学报, 40(4): 127-133.
|
| [3] |
李伟, 2019. 海洋真菌分子生态学研究概况、问题与展望[J]. 菌物学报, 38(7): 1021-1032.
|
| [4] |
凌娟, 董俊德, 张燕英, 等, 2010. 一株珊瑚礁-海草床复合生态系统固氮菌的分离与鉴定[J]. 微生物学通报, 37(7): 962-968.
|
| [5] |
汪峰,
|
| [6] |
吴宪, 胡菏, 王蕊, 等, 2022. 化肥减量和有机替代对潮土微生物群落分子生态网络的影响[J]. 土壤学报, 59(2): 545-556.
|
| [7] |
顾美英, 张志东, 唐光木, 等, 2022. 黑果枸杞不同组织内生真菌群落组成及生态功能分析[J]. 菌物学报, 41:1254-1267.
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}