
食蟹豆齿鳗线粒体基因组结构特征及系统进化分析
|
杨澜(2001—), 女, 浙江省衢州市人, 研究方向为海洋生物资源与环境。email: 3559892237@qq.com |
Editor: 林强
收稿日期: 2023-12-15
修回日期: 2024-02-11
网络出版日期: 2024-02-21
基金资助
浙江省重点研发计划项目(2021C2047)
舟山市科技计划项目(2022C41022)
国家级大学生创新创业训练计划项目(202310340056)
浙江省大学生科技创新活动计划暨新苗人才计划项目(2023R411005)
浙江海洋大学校级大学生科研创新计划项目(2023-A-025)
The mitochondrial genomic characteristics and phylogenetic relationship analysis of Pisodonophis cancrivorus (Anguilliformes, Ophichthidae)
Editor: LIN Qiang
Received date: 2023-12-15
Revised date: 2024-02-11
Online published: 2024-02-21
Supported by
Province Key Research and Development Program of Zhejiang(2021C2047)
Technology Planning Project of Zhoushan(2022C41022)
National Innovation and Entrepreneurship Training Program for College Students(202310340056)
Science and Technology Innovation Project of College Students in Zhejiang Province(2023R411005)
Innovation Project of College Students in Zhejiang Ocean University(2023-A-025)
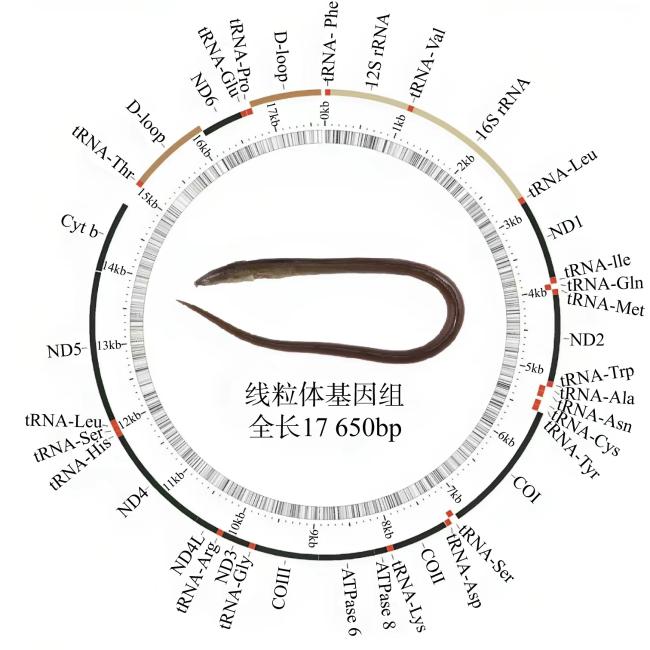
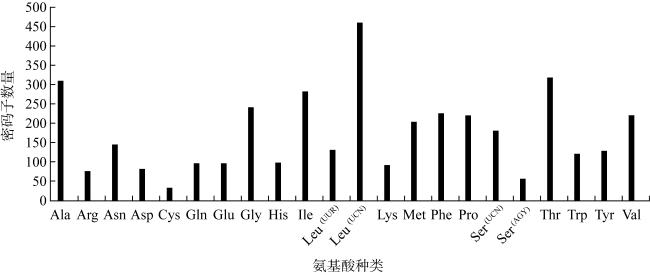
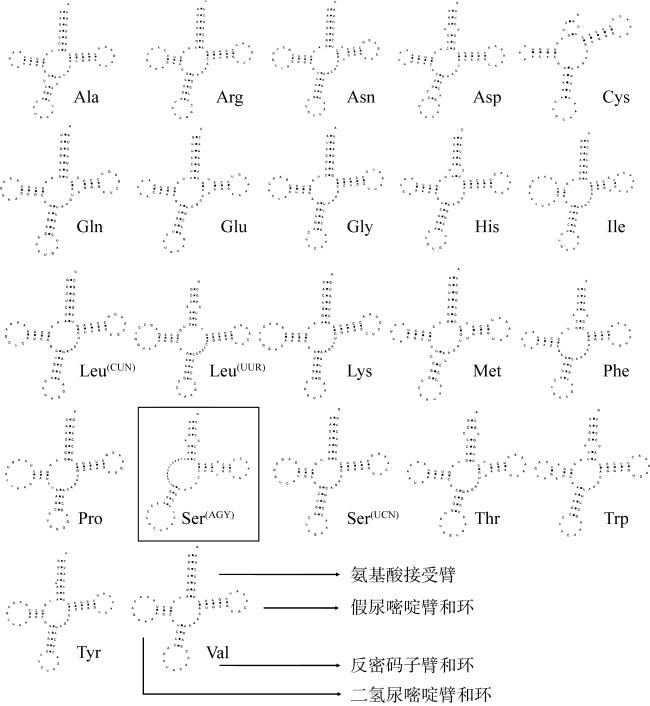
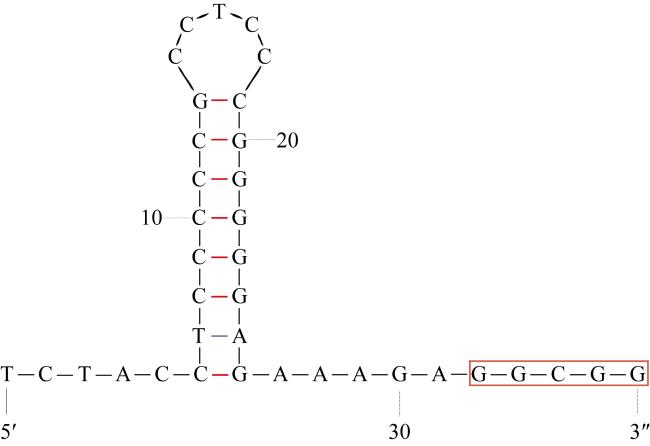
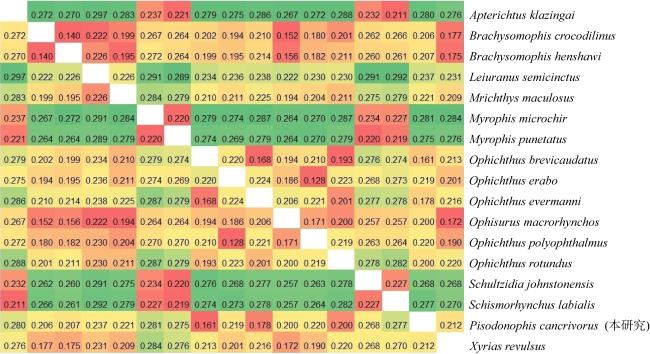
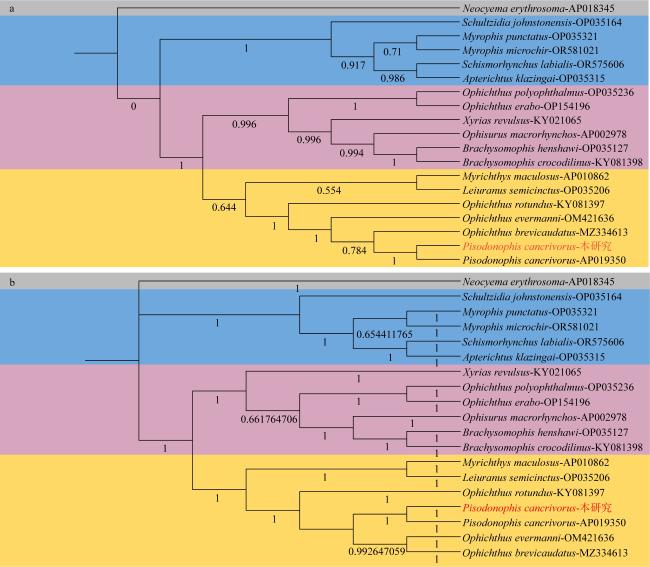
食蟹豆齿鳗(Pisodonophis cancrivorus)是我国东南沿海重要的渔获种类之一。本研究采用高通量测序技术对采自于舟山近海的食蟹豆齿鳗线粒体基因组进行了测定, 并对其结构特征及系统发育关系进行了分析。食蟹豆齿鳗线粒体DNA序列全长为17650bp, 4种碱基含量分别为A(31.72%)、G(15.58%)、T(26.26%)和C(26.44%), 表现出明显的反G偏倚和A+T富集现象。与已发表的蛇鳗科鱼类相似, 其线粒体部分基因由于自然选择的作用, 发生了串联复制-随机丢失(tandem duplication-random loss, TDRL)事件, 导致ND6基因和tRNA-Glu(E)转移到tRNA-Thr(T)和tRNA-Pro(P)之间, 并且ND6基因上游出现了另一个长度为966bp的控制区, 两个控制区呈现独立演化的特征。13个蛋白质编码基因中分别存在2种启动子(ATG、GTG)和4种终止子(TAG、TAA、TA-、T--), 且仅COI基因以GTG作为起始密码子。编码的3 824个氨基酸中, 亮氨酸(Leu)含量最高而半胱氨酸(Cys)含量最低。22种tRNA的二级结构中存在17处碱基错配, 且仅tRNA-SerAGY(S)由于缺少二氢尿嘧啶臂(DHU臂)无法折叠形成典型的三叶草结构。长度为21bp的轻链复制起始区(origin of L-strand replication region, OL)位于“WANCY”基因簇内, 其发夹型二级结构(hairpin secondary structure)的3'端具有与爬行动物类似的保守功能基序3'-GGCGG-5'。将17种蛇鳗科鱼类的线粒体12个蛋白质编码基因序列去除终止密码子之后进行串联, 基于Kimura双参数模型计算种间遗传距离为0.128~0.297。使用最大似然法(maximum likelihood, ML)和贝叶斯法(Bayesian inference, BI)构建系统发育树, 显示了蛇鳗科鱼类复杂的演化关系, 食蟹豆齿鳗位于进化树的基部, 与短尾蛇鳗(Ophichthus brevicaudatus)和艾氏蛇鳗(O.evermanni)亲缘关系最近, 是该类群中较晚发生遗传分化的种类。
杨澜 , 赵耀 , 刘玉萍 , 杨天燕 . 食蟹豆齿鳗线粒体基因组结构特征及系统进化分析[J]. 热带海洋学报, 2024 , 43(6) : 145 -159 . DOI: 10.11978/2023196
Pisodonophis cancrivorus is an important fish species in the southeast coast of China. In this study, the complete mitochondrial genome of P. cancrivorus was obtained by high-throughput sequencing technique, as well as the structural characteristics and phylogenetic relationships were also analyzed. The total length of the complete mitochondrial DNA is 17650bp, and the base compositions are A (31.72%), G (15.58%), T (26.26%) and C (26.44%), respectively, showing an obvious anti-G bias and A+T preference. Similar to the published mitogenomes of Ophichthidae species, some genes have undergone a tandem duplication-random loss (TDRL) event due to the natural selection, causing the ND6 gene and its conjoint tRNA-Glu(E) to be translocated between tRNA-Thr(T) and tRNA-Pro(P). Besides, another D-loop region with length of 966bp is located upstream of the ND6 gene, and these two D-loop regions show a trend of independent evolution. There are two start codons (ATG, GTG) and four stop codons (TAG, TAA, TA-, T--) among the 13 protein-coding genes (PCGs), and only COI gene is initiated with GTG. In the total of 3 824 encoded amino acids, the content of Leu is the highest, while the content of Cys is the lowest. There are 17 DNA mismatches in the secondary structures of 22 tRNAs. All tRNAs can form the clover-leaf structures, excepting tRNA-SerAGY(S) because of lacking the DHU arm. The origin of L-strand replication region (OL) with the length of 21bp is situated in the “WANCY” region, and the conserved motif 3'-GGCGG-5' is detected in the 3' end of its hairpin secondary structure. The sequences of 12 PCGs (without ND6) are concatenated by removing stop codons, and the interspecific genetic distances of 17 Ophichthidae fishes are calculated to be 0.128-0.297 based on the Kimura two-parameter (K2P) model. The phylogenetic trees constructed by the maximum likelihood (ML) and Bayesian inference (BI) methods show a complex evolutionary relationship of Ophichthidae fishes. P. cancrivorus is located at the base of the phylogenetic tree and closely related to Ophichthus brevicaudatus and O. evermanni, suggesting that it may diverge later than other snake eels.
表1 食蟹豆齿鳗线粒体基因组注释Tab. 1 The mitogenome annotation of P. cancrivorus |
| 基因 | 编码链 | 起始位点 | 终止位点 | 长度/bp | 基因间隔/bp | 编码氨基酸数量 | 起始密码子 | 终止密码子 | 反密码子 |
|---|---|---|---|---|---|---|---|---|---|
| tRNA-Phe(F) | H | 1 | 68 | 68 | 0 | GAA | |||
| 12S rRNA | H | 69 | 1 027 | 959 | 0 | ||||
| tRNA-Val(V) | H | 1 028 | 1 098 | 71 | 0 | TAC | |||
| 16S rRNA | H | 1 099 | 2 799 | 1 701 | 0 | ||||
| tRNA-LeuUUR(L) | H | 2 800 | 2 875 | 76 | 0 | TAA | |||
| ND1 | H | 2 876 | 3 844 | 969 | 0 | 322 | ATG | TAA | |
| tRNA-Ile(I) | H | 3 847 | 3 919 | 73 | 2 | GAT | |||
| tRNA-Gln(Q) | L | 3 919 | 3 989 | 71 | -1 | TTG | |||
| tRNA-Met(M) | H | 3 989 | 4 057 | 69 | -1 | CAT | |||
| ND2 | H | 4 058 | 5 114 | 1 057 | 0 | 352 | ATG | T-- | |
| tRNA-Trp(W) | H | 5 115 | 5 183 | 69 | 0 | TCA | |||
| tRNA-Ala(A) | L | 5 185 | 5 253 | 69 | 1 | TGC | |||
| tRNA-Asn(N) | L | 5 255 | 5 327 | 73 | 1 | GTT | |||
| OL | H | 5 329 | 5 349 | 21 | 1 | ||||
| tRNA-Cys(C) | L | 5 371 | 5 437 | 67 | 21 | GCA | |||
| tRNA-Tyr(Y) | L | 5 438 | 5 509 | 72 | 0 | GTA | |||
| COI | H | 5 511 | 7 151 | 1 641 | 1 | 546 | GTG | TAA | |
| tRNA-SerUCN(S) | L | 7 169 | 7 239 | 71 | 17 | TGA | |||
| tRNA-Asp(D) | H | 7 245 | 7 312 | 68 | 5 | GTC | |||
| COII | H | 7 319 | 8 009 | 691 | 6 | 230 | ATG | T-- | |
| tRNA-Lys(K) | H | 8 010 | 8 084 | 75 | 0 | TTT | |||
| ATP8 | H | 8 086 | 8 253 | 168 | 1 | 55 | ATG | TAA | |
| ATP6 | H | 8 244 | 8 923 | 680 | -10 | 226 | ATG | TA- | |
| COIII | H | 8 924 | 9 708 | 785 | 0 | 261 | ATG | TA- | |
| tRNA-Gly(G) | H | 9 709 | 9 780 | 72 | 0 | TCC | |||
| ND3 | H | 9 781 | 10 129 | 349 | 0 | 116 | ATG | T-- | |
| tRNA-Arg(R) | H | 10 130 | 10 199 | 70 | 0 | TCG | |||
| ND4L | H | 10 200 | 10 496 | 297 | 0 | 98 | ATG | TAA | |
| ND4 | H | 10 490 | 11 870 | 1 381 | -7 | 610 | ATG | T-- | |
| tRNA-His(H) | H | 11 871 | 11 939 | 69 | 0 | GTG | |||
| tRNA-SerAGY(S) | H | 11 940 | 12 009 | 70 | 0 | GCT | |||
| tRNA-LeuCUN(L) | H | 12 010 | 12 082 | 73 | 0 | TAG | |||
| ND5 | H | 12 083 | 13 918 | 1 836 | 0 | 611 | ATG | TAG | |
| Cyt b | H | 13 933 | 15 073 | 1 141 | 14 | 380 | ATG | T-- | |
| tRNA-Thr(T) | H | 15 074 | 15 145 | 72 | 0 | TGT | |||
| D-loop1 | H | 15 146 | 16 111 | 966 | 0 | ||||
| ND6 | L | 16 112 | 16 630 | 519 | 0 | 172 | ATG | TAG | |
| tRNA-Glu(E) | L | 16 631 | 16 699 | 69 | 0 | TTC | |||
| tRNA-Pro(P) | L | 16 703 | 16 775 | 73 | 3 | TGG | |||
| D-loop2 | H | 16 776 | 17 650 | 875 | 0 |
注: H和L代表重链和轻链; TA-和T--代表不完整终止密码子, “-”代表碱基重叠 |
表2 食蟹豆齿鳗线粒体碱基组成与偏倚Tab. 2 The base compositions and skewness in the mitogenome of P. cancrivorus |
| 基因/区域 | 长度/bp | A/% | T/% | G/% | C/% | A+T/% | AT偏倚 | GC偏倚 |
|---|---|---|---|---|---|---|---|---|
| 线粒体全序列 | 17 650 | 31.72 | 26.26 | 15.58 | 26.44 | 57.98 | 0.094 | -0.259 |
| 13个蛋白质编码基因 | 11 514 | 29.33 | 28.23 | 15.46 | 26.98 | 57.56 | 0.019 | -0.271 |
| 密码子第1位点 | 3 849 | 28.55 | 23.33 | 22.79 | 25.33 | 51.88 | 0.101 | -0.053 |
| 密码子第2位点 | 3 831 | 23.52 | 35.47 | 13.94 | 27.07 | 58.99 | -0.203 | -0.320 |
| 密码子第3位点 | 3 834 | 35.92 | 25.93 | 9.62 | 28.53 | 61.85 | 0.162 | -0.496 |
| ND1 | 969 | 27.86 | 27.86 | 15.38 | 28.90 | 55.72 | 0.000 | -0.305 |
| ND2 | 1 057 | 33.59 | 24.50 | 13.62 | 28.29 | 58.09 | 0.156 | -0.350 |
| COI | 1 641 | 28.28 | 29.25 | 17.55 | 24.92 | 57.53 | -0.017 | -0.174 |
| COII | 691 | 30.82 | 26.19 | 15.63 | 27.36 | 57.01 | 0.081 | -0.273 |
| ATP8 | 168 | 33.33 | 25.00 | 10.12 | 31.55 | 58.33 | 0.143 | -0.514 |
| ATP6 | 680 | 29.12 | 28.97 | 12.35 | 29.56 | 58.09 | 0.003 | -0.411 |
| COIII | 785 | 28.15 | 27.90 | 17.20 | 26.75 | 56.05 | 0.004 | -0.217 |
| ND3 | 349 | 28.37 | 31.52 | 14.04 | 26.07 | 59.89 | -0.053 | -0.300 |
| ND4L | 297 | 28.28 | 26.60 | 13.47 | 31.65 | 54.88 | 0.031 | -0.403 |
| ND4 | 1 381 | 29.98 | 27.59 | 13.76 | 28.67 | 57.57 | 0.042 | -0.351 |
| ND5 | 1 836 | 32.57 | 26.85 | 12.91 | 27.67 | 59.42 | 0.096 | -0.364 |
| Cyt b | 1 141 | 28.66 | 29.71 | 15.07 | 26.56 | 58.37 | -0.018 | -0.276 |
| ND6 | 519 | 38.73 | 15.03 | 14.06 | 32.18 | 53.76 | 0.441 | -0.392 |
| 12S rRNA | 959 | 33.58 | 21.38 | 20.02 | 25.02 | 54.96 | 0.222 | -0.111 |
| 16S rRNA | 1 701 | 37.92 | 19.05 | 18.99 | 24.04 | 56.97 | 0.331 | -0.117 |
| D-loop1 | 966 | 32.19 | 32.51 | 14.29 | 21.01 | 64.70 | 0.441 | -0.392 |
| D-loop2 | 875 | 36.23 | 29.60 | 10.74 | 23.43 | 65.83 | 0.441 | -0.392 |
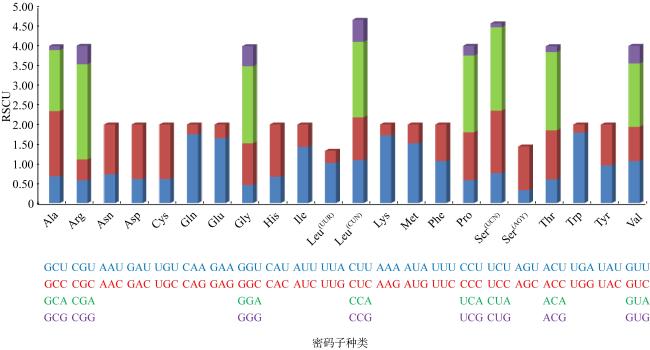
表3 13个蛋白质编码基因密码子使用频率Tab. 3 Frequency of codon usages in 13 PCGs for P. cancrivorus |
| 密码子 | 计数 | RSCU | 密码子 | 计数 | RSCU | 密码子 | 计数 | RSCU | 密码子 | 计数 | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 121 | 1.07 | UCU(S) | 30 | 0.76 | UAU(Y) | 62 | 0.96 | UGU(C) | 10 | 0.61 |
| UUC(F) | 105 | 0.93 | UCC(S) | 63 | 1.59 | UAC(Y) | 67 | 1.04 | UGC(C) | 23 | 1.39 |
| UUA(L) | 101 | 1.02 | UCA(S) | 84 | 2.12 | UAA(*) | 0 | 0.00 | UGA(W) | 108 | 1.79 |
| UUG(L) | 31 | 0.31 | UCG(S) | 4 | 0.10 | UAG(*) | 1 | 0.80 | UGG(W) | 13 | 0.21 |
| CUU(L) | 108 | 1.09 | CCU(P) | 32 | 0.58 | CAU(H) | 33 | 0.67 | CGU(R) | 11 | 0.58 |
| CUC(L) | 108 | 1.09 | CCC(P) | 67 | 1.22 | CAC(H) | 66 | 1.33 | CGC(R) | 10 | 0.53 |
| CUA(L) | 190 | 1.92 | CCA(P) | 107 | 1.95 | CAA(Q) | 84 | 1.75 | CGA(R) | 46 | 2.42 |
| CUG(L) | 55 | 0.56 | CCG(P) | 14 | 0.25 | CAG(Q) | 12 | 0.25 | CGG(R) | 9 | 0.47 |
| AUU(I) | 202 | 1.43 | ACU(T) | 48 | 0.6 | AAU(N) | 53 | 0.73 | AGU(S) | 13 | 0.33 |
| AUC(I) | 81 | 0.57 | ACC(T) | 100 | 1.25 | AAC(N) | 92 | 1.27 | AGC(S) | 44 | 1.11 |
| AUA(M) | 155 | 1.52 | ACA(T) | 159 | 1.99 | AAA(K) | 79 | 1.72 | AGA(*) | 2 | 1.60 |
| AUG(M) | 49 | 0.48 | ACG(T) | 12 | 0.15 | AAG(K) | 13 | 0.28 | AGG(*) | 2 | 1.60 |
| GUU(V) | 59 | 1.07 | GCU(A) | 53 | 0.68 | GAU(D) | 25 | 0.61 | GGU(G) | 28 | 0.46 |
| GUC(V) | 48 | 0.87 | GCC(A) | 129 | 1.66 | GAC(D) | 57 | 1.39 | GGC(G) | 64 | 1.06 |
| GUA(V) | 89 | 1.61 | GCA(A) | 120 | 1.55 | GAA(E) | 79 | 1.65 | GGA(G) | 118 | 1.96 |
| GUG(V) | 25 | 0.45 | GCG(A) | 8 | 0.10 | GAG(E) | 17 | 0.35 | GGG(G) | 31 | 0.51 |
注: 黑体表示偏好密码子 |
图6 基于K2P模型计算的蛇鳗科不同种间遗传距离Fig. 6 The genetic distances between different species of Ophichthidae based on K2P model |
| [1] |
陈慧林, 朱永航, 范海页, 等, 2023. 基于18S和28S rDNA基因的房舍甲螨DNA条形码比较研究[J]. 中国寄生虫学与寄生虫病杂志, 41(2): 163-169.
|
| [2] |
陈治, 高天翔, 2023. 线粒体12S与COI条形码对海洋鱼类的鉴定差异[J]. 海南热带海洋学院学报, 30(2): 10-16.
|
| [3] |
邓家刚, 2008. 广西海洋药物[M]. 南宁: 广西科学技术出版社.
|
| [4] |
董江星, 时伟, 孔晓瑜, 等, 2018. 褐斜鲽(Plagiopsetta glossa)线粒体基因组特征及重排机制研究[J]. 热带海洋学报, 37(1): 1-11.
|
| [5] |
龚理, 时伟, 司李真, 等, 2013. 鱼类线粒体DNA重排研究进展[J]. 动物学研究, 34(6): 666-673.
|
| [6] |
郭新红, 刘少军, 刘巧, 等, 2004. 鱼类线粒体DNA研究新进展[J]. 遗传学报, 31(9): 983-1000.
|
| [7] |
连总强, 滚双宝, 李力, 等, 2017. 基于第二代测序技术兰州鲇线粒体基因组全序列测定与分析[J]. 水生生物学报, 41(2): 334-345.
|
| [8] |
梁日深, 杨杰銮, 谢瑞琳, 等, 2022. 巨石斑鱼与斜带石斑鱼线粒体基因组测序及物种有效性分析[J]. 中国海洋大学学报, 52(6): 50-61.
|
| [9] |
林龙山, 张静, 宋普庆, 等, 2013. 东山湾及其邻近海域常见游泳动物[M]. 北京: 海洋出版社.
|
| [10] |
刘东, 2005. 中国蛇鳗科鱼类嗅觉器官的比较形态学及其系统发育研究[D]. 上海: 上海海洋大学.
|
| [11] |
刘焕章, 2004. 用mtDNA 12S rRNA序列变异检验鲤形目鱼类系统发育关系[J]. 遗传学报, 31(2): 137-142.
|
| [12] |
刘继兵, 赵洪喜, 2022. 基于18S rRNA的胎毛滴虫感染PCR诊断方法的建立[J]. 中国寄生虫学与寄生虫病杂志, 40(5): 682-685.
|
| [13] |
刘凯, 冯晓宇, 马恒甲, 等, 2020. 钱塘江三角鲂线粒体基因组测序及其结构特征分析[J]. 浙江农业学报, 32(9): 1591-1608.
|
| [14] |
刘文强, 贾玉萍, 赵宏坤, 2006. 16 S rRNA在细菌分类鉴定研究中的应用[J]. 动物医学进展, 27(11): 15-18.
|
| [15] |
刘毅敏, 赵先英, 王祥智, 等, 2006. 基于自由能的生物大分子结构研究[J]. 化学世界, 47(8): 508-510.
|
| [16] |
宁子君, 刘玉萍, 张书飞, 等, 2022. 艾氏蛇鳗线粒体基因组全序列结构分析和系统发育关系探讨[J]. 中国水产科学, 29(9): 1264-1276.
|
| [17] |
钱立富, 2018. 蛇类线粒体基因组结构演化研究及原矛头蝮属生物地理学分析[D]. 合肥: 安徽大学.
|
| [18] |
申欣, 田美, 孟学平, 等, 2014. 鳗鲡目鱼类线粒体蛋白质编码基因易位及系统演化关系分析[J]. 海洋学报, 36(4): 73-81.
|
| [19] |
唐文乔, 张春光, 2004. 蛇鳗科分类综述及中国蛇鳗科系统分类(鱼纲, 鳗鲡目)[J]. 上海水产大学学报, 13(1): 16-22.
|
| [20] |
唐优良, 章群, 余帆洋, 等, 2011. 基于12S rRNA部分序列分析的中国8种笛鲷科鱼类系统发育初探[J]. 海洋科学, 35(2): 22-26.
|
| [21] |
杨婧, 黄原, 2016. 线粒体基因组的高通量测序策略[J]. 生命科学, 28(1): 112-117.
|
| [22] |
张燕萍, 郑红梅, 邵芳, 等, 2016. 沙塘鳢线粒体基因组全序列的测定和分析[J]. 常熟理工学院学报(自然科学), 30(4): 97-104.
|
| [23] |
张稚兰, 林汝榕, 邢炳鹏, 2017. COI基因序列在蛇鳗科鱼类种类鉴定中的适用性研究[J]. 应用海洋学学报, 36(3): 411-416.
|
| [24] |
钟东, 赵贵军, 张振书, 等, 2002. 基因组内碱基分布整体均衡与局部不均衡的研究进展[J]. 遗传, 24(3): 351-355.
|
| [25] |
周慧琦, 2014. 基因组GC含量与碱基、密码子和氨基酸使用偏好的关系[D]. 成都: 电子科技大学.
|
| [26] |
邹新慧, 葛颂, 2008. 基因树冲突与系统发育基因组学研究[J]. 植物分类学报, 46(6): 795-807.
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}