
红树植物拟海桑及其亲本的根际细菌群落特征分析
|
叶锦成(1997—), 男, 广东省云浮罗定市人。email: |
Copy editor: 林强
收稿日期: 2021-08-30
修回日期: 2021-10-11
网络出版日期: 2021-10-13
基金资助
海南省林业科学研究院(海南省红树林研究院)基础性工作(KYYS-2021-04)
岭南师范学院红树林研究院开放课题(YBXM09)
海南省科研院所技术创新专项(JCYK-2021-10)
Analysis of rhizosphere bacterial community characteristics of mangrove plant Sonneratia × gulngai and its parents
Copy editor: LIN Qiang
Received date: 2021-08-30
Revised date: 2021-10-11
Online published: 2021-10-13
Supported by
The Project of Basic Scientific Research Work of Hainan Forestry Research Institute (Hainan mangrove research institute)(KYYS-2021-04)
The Open Project of Mangrove Research Institute, Lingnan Normal University(YBXM09)
The Project of Technical Innovation Special Project of Hainan Scientific Research Institutes under Grant(JCYK-2021-10)
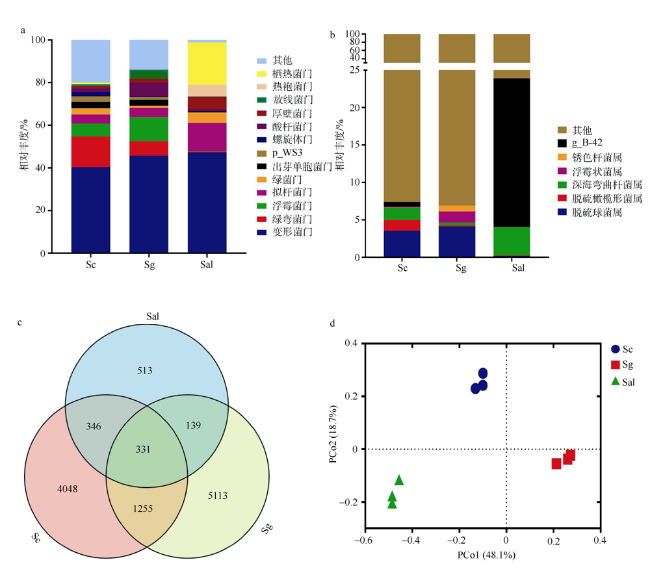
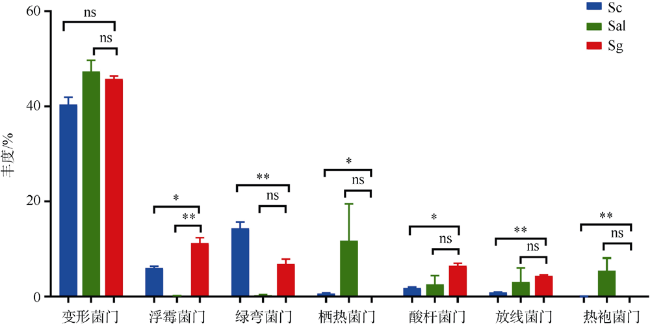
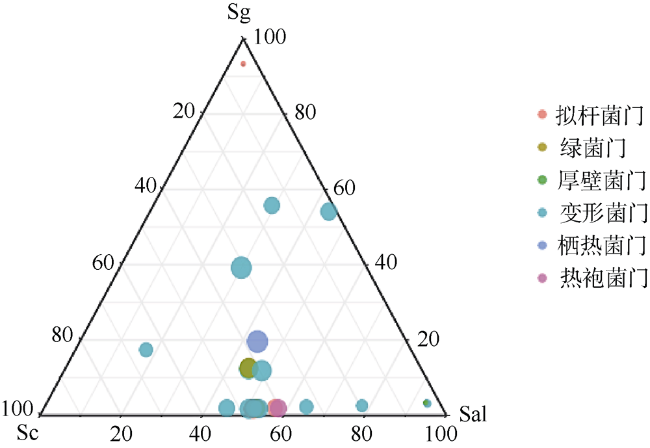
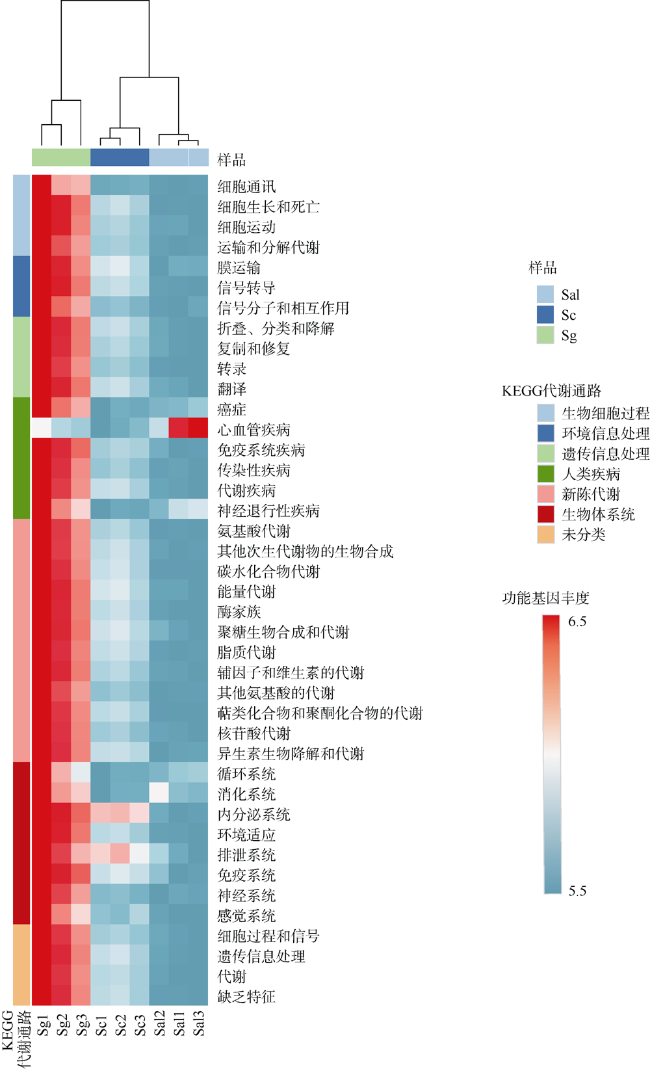
植物根际微生物群落能够从亲本传递给子代, 从而影响植物的表型。野外调查发现, 海桑属(Sonneratia)红树植物自然杂交杂种拟海桑在野外通常比亲本生长更为强壮, 表现出极强的生存优势。为探究这一现象的原因, 本研究从根际微生物角度出发, 利用细菌16S rRNA基因高通量测序技术, 对采自海南省东寨港的三种红树植物拟海桑(S. × gulngai)及其亲本海桑(S. caseolaris)和杯萼海桑(S. alba)的根际土壤进行根际细菌群落特征分析。结果表明, 三种红树植物根际细菌群落多样性高, 种类丰富, 分属于30门242科351属, 其中变形菌门(Proteobacteria)为最优势门, 在各个样本中丰度超过40%, 子代拟海桑继承亲本的根际微生物多数都属于这一类群。研究发现, 子代拟海桑与两亲本的根际细菌类群组成在门水平存在显著差异, 其中酸杆菌门(Acidobacteria)和放线菌门(Actinobacteria)在子代拟海桑中丰度分别为4.3%和6.5%, 显著高于亲本1%~2%的含量; 在亲本杯萼海桑中, 丰度高达19.8%的栖热菌门(Thermi)在子代中丰度仅有1%, 而热袍菌门(Thermotogae)(5%)甚至消失。总之, 子代拟海桑与亲本杯萼海桑的根际微生物群落组成相对于亲本海桑表现出更大的差异性。土壤理化性质分析发现, 子代拟海桑的土壤全氮(total nitrogen, TN)含量显著低于亲本, 含量相差3倍以上, 相关性分析表明, TN浓度与菌群中的浮霉菌门(Planctomycetes)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)丰度显著负相关。通过功能预测分析发现, 子代拟海桑微生物群落中与基础代谢相关的碳水化合物、氨基酸、能量以及脂质代谢相关的功能基因丰度显著都高于亲本, 表现出代谢能力的增强。本研究认为, 子代拟海桑对亲本的根际土壤微生物进行了选择性继承, 菌群组成更为合理, 在保持菌群高度多样性的同时, 一些根际促生菌的含量增加, 使菌群基础代谢能力增加, 更有利于子代拟海桑的生长。
叶锦成 , 陈毅青 , 高琳 , 周鲜娇 , 钟才荣 , 张颖 , 王芸 . 红树植物拟海桑及其亲本的根际细菌群落特征分析[J]. 热带海洋学报, 2022 , 41(6) : 75 -89 . DOI: 10.11978/2021114
Plant rhizosphere microbes can be transferred directly from mother to offspring, these vertically-transmitted organisms can affect host phenotype. Field investigation revealed that Sonneratia × gulngai, a natural hybrid of mangrove plants of the genus Sonneratia, usually grew stronger than its parents, and showed stronger survival advantage. In order to explore the reasons for this phenomenon, the offspring's microbiome composition (S. × gulngai) was compared with its parents S. caseolaris and S. alba by the bacterial 16S rRNA gene high-throughput sequencing technology. The sediment samples were collected from the Dongzhaigang Mangrove National Nature Reserve of Hainan Island. The result showed the rhizosphere bacterial communities of the three mangroves were highly diverse and rich, and distributed into 30 phyla, 242 families and 351 genera. Proteobacteria contributed to 40% of all reads and constituted the predominant members, most of the inherited rhizosphere microbiome belonged to this phylum. Significant differences were also observed at the phylum level. In offspring S. ×gulngai, the abundance of Actinobacteria and Acidobacteria was 4.3% and 6.5% respectively, which are significantly higher than parent's 1%~2%; Thermi species were enriched to be dominant populations with relative abundances of 19.8% in the parent S. alba, but the numbers was only 1% in the offspring S. × gulngai, and found Thermotogae phylum (5%) was absent in the rhizosphere bacterial community of S.× gulngai. In short, microbiome composition differed more strongly between offspring S. × gulngai and the parent S. alba than S. caseolaris. The analysis of soil physical and chemical properties found that the soil total nitrogen (TN) content of S. × gulngai was significantly lower than that of the parent, with a difference of more than 3 times. Correlation analysis showed that the concentration of TN was significantly negatively correlated with the abundance of Planctomycetes, Actinobacteria, and Acidobacteria. Through functional prediction, some functional genes related to the basic metabolism in the offspring S. × gulngai microbial community were greatly enriched, S.× gulngai's metabolic capacity was enhanced. These findings showed that the selectively inherited some the rhizosphere soil microorganisms from their parents, which made the offspring's microbiome composition more reasonable and maintained higher diversity, some rhizosphere growth-promoting bacteria particularly enriched and increased the basal metabolic capacity, these changes promoted S. × gulngai to grow better than its parent.
Key words: mangrove; Sonneratia; rhizosphere microbial; bacterial community; diversity
表1 土壤样本测序数据量统计Tab. 1 Statistics of soil samples volume sequencing data |
| 样品 | OTUs | 测序总量 |
|---|---|---|
| Sc1 | 6333 | 38105 |
| Sc2 | 5987 | 37913 |
| Sc3 | 6059 | 38142 |
| Sal1 | 1037 | 39702 |
| Sal2 | 1905 | 37969 |
| Sal3 | 1336 | 37147 |
| Sg1 | 5264 | 39259 |
| Sg2 | 6409 | 39784 |
| Sg3 | 6975 | 39264 |
注: 操作分类单元OTUs为按照97%的相似度对非重复序列(不含单序列)进行OTU聚类得到的OTU代表序列量 |
图1 各样本细菌群落构成与比较。a. 各样品菌群组成在门水平相对丰度; b. 各样品菌群组成在属水平相对丰度; c. Venn图展示不同样本的共有和特有的OTU数量; d. 基于Weighted unifrac距离的主坐标分析 Fig. 1 Composition and comparison of bacterial community in each mangrove species. (a) Relative abundance map at phylum level. (b) Relative abundance map at genus level. (c) The Venn diagram shows the number of common and unique OTUs for different samples. (d) Principal coordinate analysis generated using Weighted unifrac distance |
表2 三种海桑的根际细菌群落α多样性指数分析(平均值±标准误差)Tab. 2 Alpha diversity index of rhizosphere bacterial community of three species of Sonneratia (Mean±SD) |
| 红树物种 | 香农维纳指数 | OTUs观测量 | Chao1指数 | Ace指数 | 辛普森指数 |
|---|---|---|---|---|---|
| Sc | 10.44±0.12a | 5196.67±43.59a | 7482.27±197.88a | 7883.287±140.61a | 1±0a |
| Sg | 10.53±0.24a | 5353.33±562.37a | 6882.33±769.47a | 7231.98±801.72a | 1±0a |
| Sal | 5.65±0.56b | 1289.67±357.69b | 2229.13±464.49b | 2363.41±533.58b | 0.93±0.02b |
注: 上标不同字母的比较样本之间存在显著差异 |
表3 土壤样品的理化性质(平均值±标准误差)和ANOVA显著性分析、Duncan检验多重比较结果Tab. 3 Physicochemical characteristics of soil samples (Mean±SD) and ANOVA significance analysis, Duncan test multiple comparison results |
| 理化指标 | 样品 | ||
|---|---|---|---|
| Sc | Sg | Sal | |
| TC/(mg.g-1) | 24.33±2.07a | 13.17±0.60b | 15.60±2.30b |
| TN/(mg.g-1) | 4.33±0.06b | 1.21±0.05c | 4.64±0.04a |
| TP/(mg.g-1) | 0.34±0.02b | 0.72±0.04a | 0.67±0.03a |
| AK/(mg.g-1) | 0.51±0.01b | 0.34±0.02c | 0.67±0.01a |
| C/N | 5.62±0.52b | 10.82±0.13a | 3.36±0.52c |
| N/P | 12.45±0.67a | 1.68±0.03c | 6.93±0.24b |
注: 上标不同字母的比较样本之间存在显著差异(P<0.05) |
表4 土壤理化性质与门水平主要细菌的相关性Tab. 4 Correlation between soil physical and chemical properties and main bacteria at phylum level |
| 细菌门 | 中文名 | TC | TN | TP | AK | C/N | N/P |
|---|---|---|---|---|---|---|---|
| Proteobacteria | 变形菌门 | -0.817** | 0.217 | 0.700* | 0.159 | -0.283 | -0.667* |
| Chloroflexi | 绿弯菌门 | 0.433 | -0.467 | -0.517 | -0.402 | 0.433 | 0.467 |
| Planctomycetes | 浮霉菌门 | -0.417 | -0.917** | 0.333 | -0.854** | 0.883** | -0.433 |
| Bacteroidetes | 拟杆菌门 | -0.233 | 0.65 | -0.05 | 0.678* | -0.817** | 0.1 |
| Chlorobi | 绿菌门 | 0.35 | 0.967** | -0.267 | 0.879** | -0.933** | 0.383 |
| Gemmatimonadetes | 出芽单胞菌门 | 0.1 | -0.5 | 0.083 | -0.770* | 0.6 | -0.1 |
| WS3 | 0.417 | -0.483 | -0.533 | -0.418 | 0.417 | 0.483 | |
| Spirochaetes | 螺旋体门 | 0.433 | -0.383 | -0.55 | -0.385 | 0.35 | 0.533 |
| Acidobacteria | 酸杆菌门 | -0.433 | -0.933** | 0.3 | -0.879** | 0.850** | -0.417 |
| Firmicutes | 厚壁菌门 | -0.117 | 0.717* | 0.4 | 0.527 | -0.533 | -0.267 |
| Actinobacteria | 放线菌门 | -0.45 | -0.867** | 0.383 | -0.904** | 0.800** | -0.483 |
| Thermi | 栖热菌门 | 0.317 | 0.850** | -0.433 | 0.887** | -0.917** | 0.5 |
| Thermotogae | 热袍菌门 | 0.356 | 0.881** | -0.39 | 0.877** | -0.915** | 0.492 |
注: * P<0.05, ** P<0.01 |
| [1] |
鲍士旦, 2000. 土壤农化分析[M]. 3版. 北京: 中国农业出版社: 102-130.
|
| [2] |
曹雷雷, 田海妍, 王友绍, 等, 2015. 红树植物无瓣海桑果实的化学成分研究[J]. 热带海洋学报, 34(1): 77-82.
|
| [3] |
陈玉军, 廖宝文, 郑松发, 等, 2004. 无瓣海桑、海桑、秋茄红树人工林群落动态及物种多样性研究[J]. 应用生态学报, 15(6): 924-928.
|
| [4] |
高蕴璋, 1993. 中国海桑属小志[J]. 热带亚热带植物学报, 1(1): 11-13.
|
| [5] |
黄志强, 邱景璇, 李杰, 等, 2021. 基于16S rRNA基因测序分析微生物群落多样性[J]. 微生物学报, 61(5): 1044-1063.
|
| [6] |
江睿, 吴云超, 陈丕茂, 2021. 珠江口淇澳岛红树林湿地沉积物碳、氮分布研究[J]. 南方水产科学, 17(1): 1-9.
|
| [7] |
李海生, 2003. 中国海桑属红树植物遗传多样性研究[D]. 广州: 中山大学:11-17.
|
| [8] |
李晴晴, 徐松, 赵维, 等, 2019. 根际微生物组介导的解淀粉芽孢杆菌FH-1对水稻的促生机制[J]. 微生物学报, 59(12): 2410-2426.
|
| [9] |
李诗川, 李妮亚, 刘强, 等, 2014. 海桑属红树植物离子积累、光合和抗氧化能力及相关性分析[J]. 植物资源与环境学报, 23(3): 15-23.
|
| [10] |
林婉奇, 薛立, 2020. 基于BIOLOG技术分析氮沉降和降水对土壤微生物功能多样性的影响[J]. 生态学报, 40(12): 4188-4197.
|
| [11] |
刘京伟, 李香真, 姚敏杰, 2021. 植物根际微生物群落构建的研究进展[J]. 微生物学报, 61(2): 231-248.
|
| [12] |
柳旭, 2019. 云南省三种入侵植物根际土壤细菌群落多样性研究[D]. 保定: 河北大学: 1-45.
|
| [13] |
毛礼米, 李妮娅, 王东, 等, 2009. 海桑属6种植物花粉形态兼化石花粉指南[J]. 古生物学报, 48(2): 254-267.
|
| [14] |
齐璐, 2019. 寒旱区城市湖泊冰封期细菌群落结构特征变化研究[D]. 包头: 内蒙古科技大学: 1-42.
|
| [15] |
任健, 阎冰, 洪葵, 2012. 海南东寨港红树林不同植被土壤微生物群落结构比较[J]. 微生物学报, 52(6): 736-743.
|
| [16] |
沈芳芳, 刘影, 罗昌泰, 等, 2019. 陆地生态系统植物和土壤微生物群落多样性对全球变化的响应与适应研究进展[J]. 生态环境学报, 28(10): 2129-2140.
|
| [17] |
王光华, 刘俊杰, 于镇华, 等, 2016. 土壤酸杆菌门细菌生态学研究进展[J]. 生物技术通报, 32(2): 14-20.
|
| [18] |
王瑞江, 陈忠毅, 陈二英, 等, 1999. 国产海桑属植物的两个杂交种[J]. 广西植物, 19(3): 199-204.
|
| [19] |
王勇忠, 2015. 福建沿海滩涂底泥细菌群落结构和功能对生物修复的响应[D]. 厦门: 集美大学: 1-57.
|
| [20] |
吴冬梅, 2018. 长江口和东海海域细菌群落结构及其生态功能预测[D]. 舟山: 浙江海洋大学: 1-58.
|
| [21] |
颜栋美, 王伟, 李蜜, 等, 2018. 茅尾海无瓣海桑根际土壤细菌多样性及抑菌活性分析[J]. 南方农业学报, 49(6): 1095-1101.
|
| [22] |
闫绍鹏, 杨瑞华, 冷淑娇, 等, 2012. 高通量测序技术及其在农业科学研究中的应用[J]. 中国农学通报, 28(30): 171-176.
|
| [23] |
袁颖红, 樊后保, 刘文飞, 等, 2013. 模拟氮沉降对杉木人工林(Cunninghamia lanceolata)土壤酶活性及微生物群落功能多样性的影响[J]. 土壤, 45(1): 120-128.
|
| [24] |
张福锁, 曹一平, 1992. 根际动态过程与植物营养[J]. 土壤学报, 29(3): 239-250.
|
| [25] |
张起畅, 张文飞, 殷浩能, 等, 2020. 宏基因组测序分析东寨港红树林淤泥和水体微生物的多样性[J]. 基因组学与应用生物学, 39(1): 116-122.
|
| [26] |
张威, 刘宁, 吕慧捷, 等, 2009. TruSpec CN元素分析仪测定土壤中碳氮方法研究[J]. 分析仪器, (3): 46-49.
|
| [27] |
周卫国, 丁德文, 凌娟, 等, 2019. 海草根际微生物与海草植株的互作效应[J]. 微生物学报, 59(11): 2117-2129.
|
| [28] |
周晓光, 任鲁风, 李运涛, 等, 2010. 下一代测序技术: 技术回顾与展望[J]. 中国科学: 生命科学, 40(1): 23-37.
|
| [29] |
朱丽霞, 章家恩, 刘文高, 2003. 根系分泌物与根际微生物相互作用研究综述[J]. 生态环境, 12(1): 102-105.
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}