
Journal of Tropical Oceanography >
Discovery and verification of SNP in Acanthopagrus latus
Copy editor: YIN Bo
Received date: 2022-05-13
Revised date: 2022-07-09
Online published: 2022-07-14
Supported by
National Key R&D Program of China(2018YFD0901404)
Evaluation of the Release Effect of Acanthopagrus latus in Xiamen Bay(S20166)
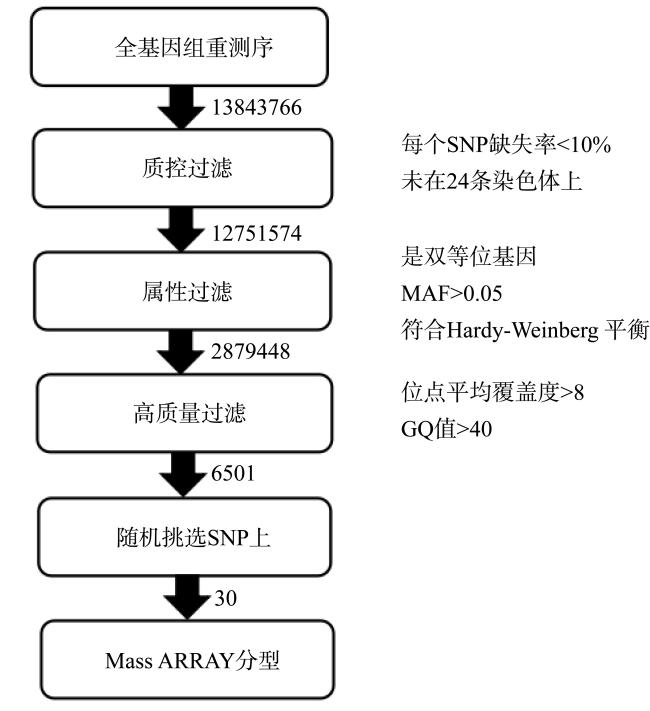
In this experiment, 'Acanthopagrus latus', an important economic fish in the southeast coast of China, was used as the experimental material. The SNPs were discovered by DNA re-sequencing in fifty A. latus, and partial SNPs were genotyped using MassARRAY® DNA mass spectrometry. The results are presented as followed: 1) the re-sequencing of 50 wild A. latus generated a total of about 233.48 GB raw data. After filtering adapters and low-quality data, 233.43 Gb clear data were obtained. The average data size of each sample was 4.67 GB, and the average GC content was 42.85%, Q20 is above 96.56%, Q30 is above 91.1%, and the comparison rate between the clear data and the reference genome is 98.06% ~ 99.47%; 2) a total of 13843766 SNPs were discovered from 50 individuals by GATK, and 6501 high-quality SNPs were obtained after filtering; 3) thirty SNPs from the high-quality SNP were selected randomly and genotyped using MassARRAY technology. The detection rate (loci that can be genotyped) was reached at 98%. The consistency between the genome re-sequencing and the MassARRAY results was 64.83%, which indicated that the two techniques were different in detecting SNPs. In summary, a method for mining, filtering and validating SNP markers of A. latus bream has been established in this study, and the developed SNP loci can be used in evaluation of proliferation and stocking effect and genome selection breeding of A. latus in the future.
Key words: Acanthopagrus latus; Re-sequencing; SNP; MassARRAY
ZHENG Guobin , ZHAO Hongbo , HUANG Liangmin , ZHANG Jing , LIU Xiande . Discovery and verification of SNP in Acanthopagrus latus[J]. Journal of Tropical Oceanography, 2023 , 42(2) : 78 -86 . DOI: 10.11978/2022108
表1 黄鳍棘鲷样品信息Tab. 1 Sample information of Acanthopagrus latus |
| 取样地 | 数量/尾 | 体长/cm | 体重/g | 体厚/cm |
|---|---|---|---|---|
| 厦门近海海区 | 37 | 20.52 ± 0.10 | 190.15 ± 5.92 | 7.27 ± 0.05 |
| 漳州东山岛海区 | 13 | 21.23 ± 0.29 | 212.53 ± 6.19 | 7.52 ± 0.08 |
图1 黄鳍棘鲷SNP标记开发和验证的流程图箭头右侧数字代表每次分析后剩余SNP数, 黑色方框右侧是数据分析的过滤参数 Fig. 1 Flow chart of SNP marker discovery and validation in Acanthopagrus latus. The remaining number of SNP after each step is listed next to the black arrows. Filter parameters used for data mining are given on the right side of black boxes |
表2 重测序数据质量统计结果Tab. 2 The results of re-sequence data quality |
| 样品 | 原始数据/Gb | 过滤数据/Gb | 比对率/% | 测序深度/× | Q20/% | Q30/% | GC含量/% | 样品 | 原始数据/Gb | 过滤数据/Gb | 比对率/% | 测序深度/× | Q20/% | Q30/% | GC含量/% |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y01 | 4.71 | 4.71 | 95.90 | 6.15 | 97.34 | 92.78 | 42.69 | Y26 | 4.79 | 4.79 | 97.00 | 6.33 | 97.33 | 92.77 | 42.80 |
| Y02 | 5.00 | 4.99 | 95.49 | 6.52 | 97.39 | 92.88 | 42.69 | Y27 | 4.59 | 4.59 | 96.96 | 6.09 | 97.27 | 92.52 | 42.93 |
| Y03 | 4.76 | 4.76 | 96.20 | 6.22 | 97.43 | 92.94 | 42.61 | Y28 | 4.63 | 4.63 | 96.76 | 6.10 | 96.56 | 91.10 | 42.78 |
| Y04 | 4.55 | 4.55 | 96.83 | 6.03 | 97.49 | 93.13 | 42.73 | Y29 | 4.74 | 4.74 | 96.74 | 6.27 | 97.34 | 92.84 | 42.83 |
| Y05 | 4.58 | 4.58 | 96.38 | 6.06 | 97.43 | 93.03 | 42.76 | Y30 | 4.48 | 4.48 | 96.96 | 5.96 | 97.54 | 93.17 | 42.66 |
| Y06 | 4.46 | 4.46 | 96.46 | 5.92 | 97.44 | 93.02 | 42.69 | Y31 | 4.85 | 4.85 | 97.2 | 6.41 | 97.47 | 93.06 | 42.66 |
| Y07 | 4.71 | 4.71 | 97.03 | 6.25 | 97.45 | 93.06 | 42.76 | Y32 | 4.17 | 4.17 | 97.10 | 5.59 | 97.26 | 92.60 | 42.75 |
| Y08 | 4.79 | 4.79 | 96.69 | 6.32 | 97.21 | 92.52 | 42.80 | Y33 | 4.98 | 4.98 | 97.27 | 6.55 | 97.27 | 92.68 | 42.99 |
| Y09 | 4.86 | 4.86 | 97.36 | 6.43 | 97.34 | 92.82 | 43.18 | Y34 | 3.69 | 3.69 | 96.8 | 5.00 | 97.29 | 92.64 | 42.96 |
| Y10 | 4.78 | 4.77 | 96.60 | 6.30 | 97.19 | 92.46 | 42.87 | Y35 | 4.92 | 4.92 | 97.33 | 6.48 | 97.12 | 92.31 | 43.03 |
| Y11 | 4.43 | 4.43 | 96.56 | 5.88 | 96.97 | 92.00 | 42.83 | Y36 | 4.86 | 4.86 | 97.64 | 6.41 | 97.23 | 92.58 | 42.95 |
| Y12 | 4.59 | 4.59 | 96.60 | 6.06 | 97.09 | 92.23 | 42.95 | Y37 | 4.88 | 4.88 | 97.32 | 6.45 | 97.04 | 92.13 | 42.79 |
| Y13 | 4.63 | 4.63 | 96.87 | 6.13 | 97.26 | 92.64 | 42.78 | Y38 | 4.84 | 4.84 | 96.84 | 6.35 | 97.25 | 92.59 | 42.74 |
| Y14 | 4.82 | 4.82 | 96.55 | 6.32 | 97.08 | 92.24 | 42.71 | Y39 | 4.59 | 4.59 | 96.43 | 6.07 | 97.28 | 92.65 | 42.85 |
| Y15 | 4.63 | 4.63 | 96.57 | 6.12 | 97.22 | 92.49 | 42.77 | Y40 | 4.23 | 4.23 | 97.54 | 5.66 | 97.36 | 92.82 | 42.95 |
| Y16 | 4.70 | 4.69 | 96.80 | 6.20 | 97.24 | 92.61 | 42.97 | Y41 | 4.73 | 4.73 | 97.65 | 6.26 | 97.49 | 93.13 | 43.01 |
| Y17 | 4.93 | 4.93 | 96.95 | 6.50 | 97.17 | 92.43 | 42.91 | Y42 | 4.37 | 4.37 | 96.58 | 5.77 | 97.26 | 92.61 | 43.08 |
| Y18 | 4.33 | 4.33 | 97.05 | 5.79 | 97.10 | 92.22 | 42.81 | Y43 | 4.90 | 4.90 | 97.08 | 6.45 | 97.42 | 92.99 | 43.01 |
| Y19 | 4.85 | 4.85 | 96.91 | 6.40 | 97.25 | 92.59 | 42.95 | Y44 | 4.82 | 4.81 | 97.36 | 6.36 | 97.49 | 93.09 | 42.99 |
| Y20 | 4.52 | 4.52 | 96.45 | 5.96 | 97.33 | 92.76 | 42.75 | Y45 | 4.95 | 4.95 | 97.14 | 6.52 | 97.50 | 93.16 | 43.02 |
| Y21 | 4.79 | 4.79 | 97.00 | 6.32 | 97.20 | 92.46 | 42.73 | Y46 | 4.61 | 4.61 | 97.09 | 5.85 | 97.46 | 93.06 | 42.68 |
| Y22 | 4.84 | 4.84 | 97.00 | 6.36 | 97.54 | 93.21 | 42.54 | Y47 | 4.75 | 4.75 | 96.72 | 6.27 | 97.42 | 92.98 | 42.86 |
| Y23 | 4.71 | 4.71 | 96.41 | 6.17 | 96.71 | 91.43 | 42.72 | Y48 | 4.50 | 4.50 | 96.85 | 5.96 | 97.44 | 93.02 | 42.90 |
| Y24 | 4.58 | 4.58 | 96.84 | 6.08 | 97.25 | 92.56 | 42.75 | Y49 | 4.42 | 4.42 | 97.43 | 5.90 | 97.17 | 92.46 | 43.33 |
| Y25 | 4.75 | 4.75 | 97.22 | 6.27 | 97.33 | 92.74 | 42.80 | Y50 | 4.88 | 4.88 | 97.36 | 6.44 | 97.38 | 92.88 | 43.03 |
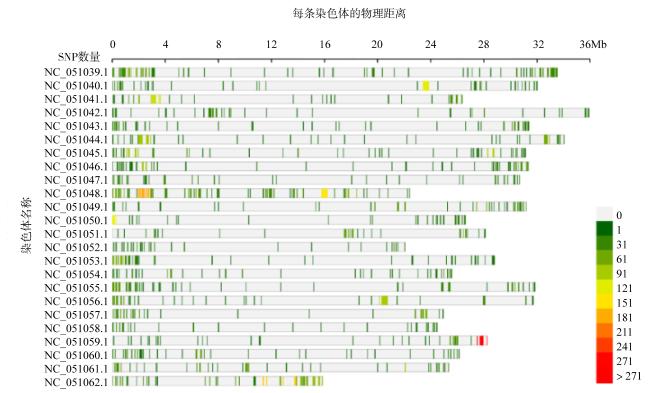
表3 基于基因组重测序黄鳍棘鲷SNP标记过滤结果Tab. 3 The results of SNP discovery and filtering in re-sequenced Acanthopagrus latus genome |
| 染色体名称 | SNP初始数 | 质控过滤后SNP数 | 属性过滤后SNP数 | 高质量过滤后SNP数 |
|---|---|---|---|---|
| NC_051039.1 | 692976 | 632996 | 144274 | 364 |
| NC_051040.1 | 646881 | 595158 | 134036 | 221 |
| NC_051041.1 | 566757 | 522152 | 117157 | 325 |
| NC_051042.1 | 690338 | 640940 | 142536 | 172 |
| NC_051043.1 | 606086 | 565937 | 129160 | 157 |
| NC_051044.1 | 697405 | 638444 | 144000 | 265 |
| NC_051045.1 | 589449 | 548080 | 124463 | 366 |
| NC_051046.1 | 615425 | 574376 | 130360 | 228 |
| NC_051047.1 | 622596 | 577032 | 131271 | 203 |
| NC_051048.1 | 476295 | 405575 | 97778 | 820 |
| NC_051049.1 | 629215 | 586030 | 132799 | 263 |
| NC_051050.1 | 574454 | 532521 | 120384 | 237 |
| NC_051051.1 | 567923 | 530390 | 119320 | 139 |
| NC_051052.1 | 462308 | 429823 | 94073 | 109 |
| NC_051053.1 | 538049 | 501748 | 110871 | 172 |
| NC_051054.1 | 535520 | 495987 | 112649 | 173 |
| NC_051055.1 | 613575 | 567469 | 128275 | 221 |
| NC_051056.1 | 669814 | 618189 | 137376 | 256 |
| NC_051057.1 | 520747 | 477109 | 104765 | 187 |
| NC_051058.1 | 509767 | 475642 | 106603 | 97 |
| NC_051059.1 | 583650 | 535046 | 121219 | 370 |
| NC_051060.1 | 563226 | 523705 | 117774 | 169 |
| NC_051061.1 | 518795 | 479194 | 110565 | 299 |
| NC_051062.1 | 329951 | 298031 | 67740 | 688 |
| 未知序列 | 22564 | 0 | 0 | 0 |
| 合计 | 13843766 | 12751574 | 2879448 | 6501 |
表4 高质量SNP多态性参数Tab. 4 Polymorphism parameters of high-quality SNPs |
| 多态性参数 | 最大值 | 最小值 | 平均值 |
|---|---|---|---|
| 观测杂合度(Ho) | 0.966 | 0.034 | 0.499 |
| 期望杂合度(He) | 0.600 | 0.034 | 0.376 |
| 多态信息含量(PIC) | 0.375 | 0.033 | 0.279 |
表5 基因组重测序与MassARRAY分型结果比较Tab. 5 Genotyping comparison between genome re-sequencing and MassARRAY |
| SNP | 染色体名称 | 位置 | MassARRAY分型检测 | 全基因组重测序 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 检测结果 | 检出率/% | MF | MAF | 检测结果 | 检出率/% | MF | MAF | |||
| Shq_178_1 | NC_051039.1 | 33527645 | CC、CG | 85 | 0.85 | 0.15 | CC、CG | 95 | 0.74 | 0.26 |
| Shq_178_2 | NC_051040.1 | 32016498 | TT | 100 | 1.00 | 0 | TT、TG | 100 | 0.88 | 0.13 |
| Shq_178_3 | NC_051041.1 | 20806265 | TT、TG | 100 | 0.95 | 0.05 | TT、TG | 100 | 0.75 | 0.25 |
| Shq_178_6 | NC_051042.1 | 30329 | AA、AG | 90 | 0.69 | 0.31 | AA、AG | 95 | 0.63 | 0.37 |
| Shq_178_8 | NC_051042.1 | 7311706 | TT、TC | 100 | 0.95 | 0.05 | TT、TC | 100 | 0.95 | 0.05 |
| Shq_178_19 | NC_051048.1 | 22404460 | AA | 95 | 1.00 | 0 | AA、AG | 90 | 0.92 | 0.08 |
| Shq_178_26 | NC_051050.1 | 34885 | CC | 95 | 1.00 | 0 | CC、CA | 95 | 0.92 | 0.08 |
| Shq_178_39 | NC_051050.1 | 38757 | GG | 100 | 1.00 | 0 | GG、GA | 90 | 0.94 | 0.06 |
| Shq_178_40 | NC_051050.1 | 42037 | TT | 100 | 1.00 | 0 | TT、TC | 95 | 0.87 | 0.13 |
| Shq_178_48 | NC_051050.1 | 59154 | TT、TC | 85 | 0.91 | 0.09 | TT、TC | 100 | 0.65 | 0.35 |
| Shq_178_60 | NC_051050.1 | 74750 | GG | 100 | 1.00 | 0 | GG、GA | 90 | 0.94 | 0.06 |
| Shq_178_63 | NC_051050.1 | 75488 | GG | 100 | 1.00 | 0 | GG、GC | 95 | 0.95 | 0.05 |
| Shq_178_65 | NC_051050.1 | 76081 | AA | 100 | 1.00 | 0 | AA、AC | 85 | 0.88 | 0.12 |
| Shq_178_70 | NC_051050.1 | 87063 | GG | 100 | 1.00 | 0 | GG、GA | 90 | 0.89 | 0.11 |
| Shq_178_72 | NC_051050.1 | 93481 | CC、CT | 100 | 0.98 | 0.03 | CC、CT | 100 | 0.95 | 0.05 |
| Shq_178_73 | NC_051050.1 | 96282 | CC | 100 | 1.00 | 0 | CC、CA | 95 | 0.82 | 0.18 |
| Shq_178_77 | NC_051050.1 | 101791 | CC | 100 | 1.00 | 0 | CC、CT | 80 | 0.94 | 0.06 |
| Shq_178_86 | NC_051050.1 | 106638 | GG、GA | 100 | 0.97 | 0.03 | GG、GA | 80 | 0.97 | 0.03 |
| Shq_178_92 | NC_051050.1 | 128900 | AA | 100 | 1.00 | 0 | AA、AG | 100 | 0.93 | 0.08 |
| Shq_178_96 | NC_051050.1 | 142395 | GG | 100 | 1.00 | 0 | GG、GA | 90 | 0.92 | 0.08 |
| Shq_178_104 | NC_051052.1 | 1588997 | AA | 100 | 1.00 | 0 | AA、AC | 100 | 0.75 | 0.25 |
| Shq_178_112 | NC_051053.1 | 1538 | GG | 100 | 1.00 | 0 | AA、AG、GG | 95 | 0.53 | 0.47 |
| Shq_178_138 | NC_051053.1 | 8936396 | AA | 100 | 1.00 | 0 | AA、AG | 95 | 0.76 | 0.24 |
| Shq_178_149 | NC_051056.1 | 31720087 | CC、CT | 100 | 0.95 | 0.05 | CC、CT | 90 | 0.86 | 0.14 |
| Shq_178_150 | NC_051056.1 | 31723482 | TT | 100 | 1.00 | 0 | TT、TC | 95 | 0.89 | 0.11 |
| Shq_178_157 | NC_051058.1 | 24478462 | AA、AG | 100 | 0.95 | 0.05 | AA、AC | 100 | 0.95 | 0.05 |
| Shq_178_158 | NC_051058.1 | 24478507 | CC、CT | 95 | 0.97 | 0.03 | CC、CT | 100 | 0.95 | 0.05 |
| Shq_178_171 | NC_051060.1 | 26149536 | GG | 100 | 1.00 | 0 | GG、GC | 100 | 0.58 | 0.43 |
| Shq_178_172 | NC_051061.1 | 336649 | TT | 100 | 1.00 | 0 | TT、TG | 100 | 0.55 | 0.45 |
| Shq_178_173 | NC_051061.1 | 22105514 | GG | 100 | 1.00 | 0 | GG、GA | 100 | 0.55 | 0.45 |
注: MF为主要等位基因频率(major allele frequency); MAF为次等位基因频率(minor allele frequency) |
| [1] |
江兴龙, 黄永春, 黄良敏, 等, 2013. 厦门湾黄鳍鲷增殖放流效果的评估[J]. 集美大学学报(自然科学版), 18(3): 161-166.
|
| [2] |
吴利娜, 张凝鋆, 孙松, 等, 2021. 微卫星分子标记技术在大黄鱼增殖放流效果评估中的应用[J]. 中国水产科学, 28(9): 1100-1108.
|
| [3] |
杨习文, 刘熠, 薛向平, 等, 2020. 基于微卫星标记的长江江苏段鲢(Hypophthalmichthys molitrix)增殖放流资源贡献率的评估[J]. 湖泊科学, 32(4): 1154-1164.
|
| [4] |
赵雨, 2021. 基于微卫星标记的日本对虾增殖放流效果评价及群体遗传学研究[D]. 天津农学院.
|
| [5] |
朱克诚, 宋岭, 刘宝锁, 等, 2020. 黄鳍棘鲷家系亲缘关系鉴定[J]. 水产学报, 44(3): 351-357.
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}