
Journal of Tropical Oceanography >
Design, synthesis and cytotoxicity of novel glycosyl-2, 5-diketopiperazine derivatives
Copy editor: LIN Qiang
Received date: 2022-06-21
Revised date: 2022-08-01
Online published: 2022-08-18
Supported by
National Natural Science Foundation of China(82073762)
Guangdong Basic and Applied Basic Research Foundation(2020A1515011045)
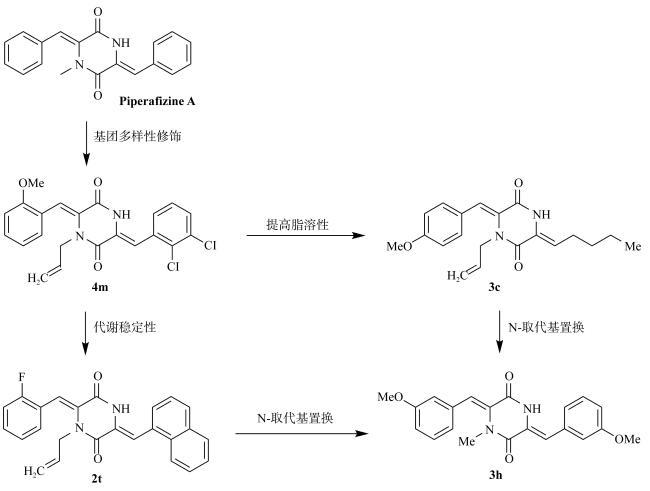
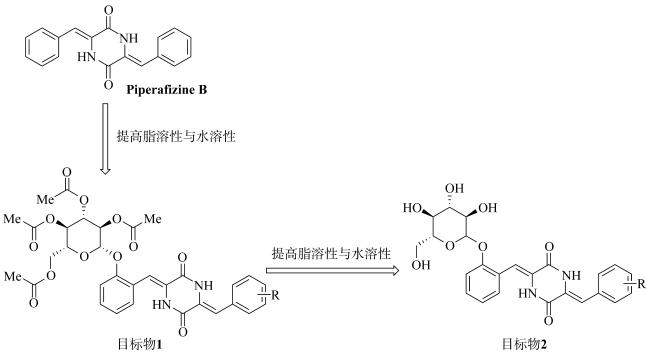
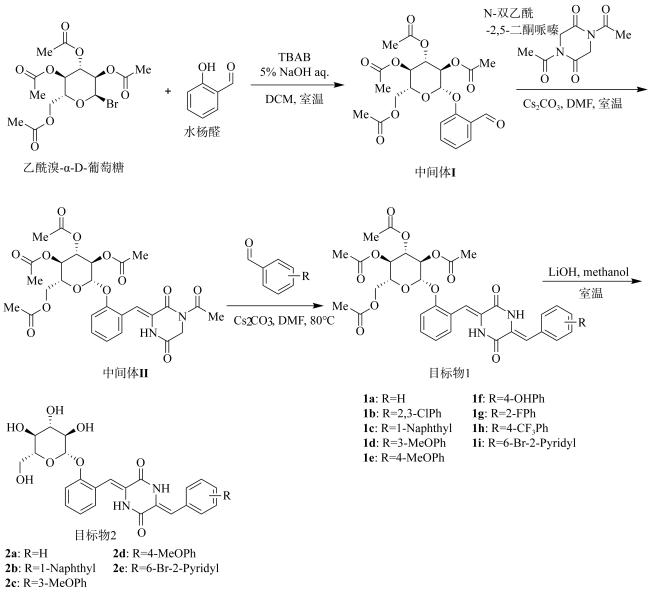
Exploring the cytotoxic compounds from marine natural products is one of the most important tasks for the discovery of anticancer agent in recent years. In this study, based on the marine natural product Piperafizine B, fourteen novel glycosyl-2, 5-diketopiperazine derivatives (1a-i, 2a-f) have been designed and synthesized by the glycosylation on the side phenol group at the 6-position of the 2, 5-diketopiperizine ring. The solubilities of the derivatives are obviously improved in both DMSO (dimethyl sulfoxide, 1.5~27.9mg·mL-1) and phosphate buffer (0.1~0.9mg·mL-1, pH=7.4). The cytotoxicity results show that some of the derivatives present good activities with the IC50 values ranging from 1.0~9.1μmol·L-1, and exhibit selective inhibitory activities on Huh-7 cell lines. Among the derivatives, compound 1c has the best inhibitory abilities on the cell lines K562, A549 and Huh-7 with an IC50 values at 1.0, 3.6 and 3.3μmol·L-1, respectively, while compounds 2a-f have no observable cytotoxicity.
LIAO Shengrong , XU Huayan , LI Xiaolin , LIU Yonghong . Design, synthesis and cytotoxicity of novel glycosyl-2, 5-diketopiperazine derivatives[J]. Journal of Tropical Oceanography, 2023 , 42(3) : 174 -185 . DOI: 10.11978/2022140
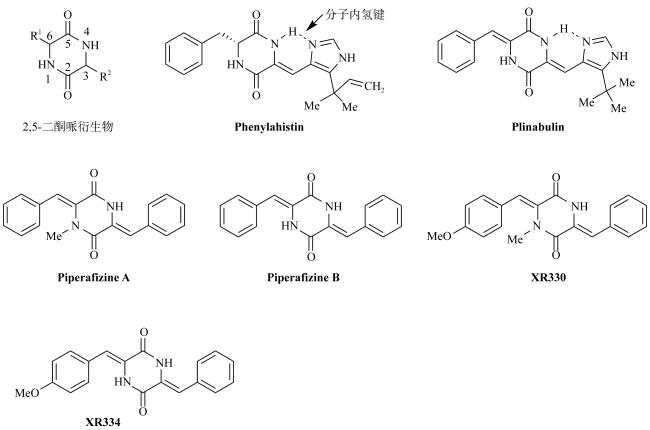
图1 2, 5-二酮哌嗪及其海洋来源的天然产物与结构衍生物PlinabulinFig. 1 2, 5-Diketopiperafizine, its marine originated natural products and the modified compound Plinabulin |
表1 化合物的溶解度Tab. 1 The solubilities of the synthesized derivatives |
| 化合物 | 溶解度/(mg·mL-1) | |
|---|---|---|
| CDMSO | C缓冲液 | |
| 1a | 21.5 | 0.9 |
| 1b | 11.8 | 0.4 |
| 1c | 13.9 | 0.2 |
| 1d | 17.0 | 0.9 |
| 1e | 12.2 | 0.5 |
| 1f | 18.0 | 0.6 |
| 1g | 21.2 | 0.8 |
| 1h | 17.6 | 0.5 |
| 1i | 27.9 | 0.2 |
| 2a | 18.3 | 0.6 |
| 2b | 12.6 | 0.1 |
| 2c | 5.70 | 0.3 |
| 2d | 7.20 | 0.8 |
| 2e | 1.50 | 0.2 |
| Piperafizine B | 0.10 | 0.05 |
表2 化合物的细胞毒活性Tab. 2 The cytotoxicities of the synthesized derivatives |
| 化合物 | IC50/(μmol·mL-1) | ||
|---|---|---|---|
| K562 | A549 | Huh-7 | |
| 1a | 4.5±0.8 | 5.2±0.6 | 3.9±0.9 |
| 1b | >10 | >10 | >10 |
| 1c | 1.0±0.08 | 3.6±0.2 | 3.3±0.4 |
| 1d | >10 | >10 | >10 |
| 1e | >10 | >10 | >10 |
| 1f | >10 | >10 | 4.2±0.6 |
| 1g | >10 | 2.9±0.2 | 3.6±0.6 |
| 1h | >10 | >10 | >10 |
| 1i | 9.1±1.0 | >10 | 4.5±0.5 |
| 2a-e | >10 | >10 | >10 |
| Piperafizine B | >10 | >10 | >10 |
| Taxol | 0.003±0.0002 | 0.002±0.0008 | 0.007±0.0005 |
| [1] |
汤勇, 廖升荣, 李晋昇, 等, 2016. 含氟2, 5-二酮哌嗪衍生物的设计、合成及细胞毒活性[J]. 中国药科大学学报, 47(4): 412-421.
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
VAN DE WATERBEEMD H,
|
| [27] |
|
| [28] |
|
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}