
Journal of Tropical Oceanography >
Characteristic analysis of the complete mitogenome of Plagiopsetta glossa and a possible mechanism for gene rearrangement
Received date: 2017-04-11
Request revised date: 2017-05-25
Online published: 2018-02-02
Supported by
National Natural Science Foundation of China (31471979, 30870283)
Copyright
The family Samaridae in Pleuronectiformes consists of three genera, namely, Plagiopsetta, Samariscus and Samaris. Studies on mitochondrial genomes of Samaris cristatus and Samariscus latus showed that there exist differences in genomic rearrangement and gene number between this two species. To determine whether there are structural differences in the species of the genus Plagiopsetta, Plagiopsetta glossa were used as the representative species for this study, and the mitochondrial genomic characteristics of this species were compared with those of Samariscus and Samaris. The length of its mitogenome is 18,723 bp, and contains 39 genes, including 13 protein-coding genes, two rRNA genes, 24 tRNA genes, two control regions, one light strand replication origin, and 13 more interspaces. Compared with the typical mitogenome in teleosts, P. glossa and S. latus have two more genes of tRNA-Cys and tRNA-Tyr (S. cristatus only one more tRNA-Cys). Additionally, each of the three flatfish has an extra control region, but the gene orders of P. glossa and S. latus mitogenomes are the same. Six genes (tRNA-Cys1, tRNA-Tyr1, tRNA-Ser1, tRNA-Lys, tRNA-Arg, and tRNA-Ser2) from different locations are clustered together forming a gene cluster, and following by this genes cluster was ND5-ND6-Glu-Cytb-Thr, all those 11 genes have no gene order change in terms of typical mitogenome.
The model of Double Replications and Random Loss was used to analyze the possible rearrangement mechanism in P. glossa mitogenome. The characteristics of gene number, gene order and 13 more interspaces provided new evidence to support the applicability of this model. The results of this study not only enrich our scientific knowledge of mitogenomic features, but also provide more data for further study on mitochondrial evolution and phylogenetic analysis for flatfish.
Key words: Pleuronectiformes; Samaridae; mitogenome; gene rearrangement; molecular evolution
DONG Jiangxing , SHI Wei , KONG Xiaoyu , CHEN Shixi . Characteristic analysis of the complete mitogenome of Plagiopsetta glossa and a possible mechanism for gene rearrangement[J]. Journal of Tropical Oceanography, 2018 , 37(1) : 1 -11 . DOI: 10.11978/2017041
Tab. 1 The primers used for amplification in Plagiopsetta glossa mitogenomes表1 扩增褐斜鲽线粒体全基因引物使用表 |
| 正向引物 | 序列(5′—3′) | 反向引物 | 序列(5′—3′) |
|---|---|---|---|
| Z15 | ATTAAAGCATAACHCTGAAGATGTTAAGAT | F2753 | TAGATAGAAACTGACCTGGATTACTCCGGT |
| Z2625 | GTTTACGACCTCGATGTTGGATCAGGACAT | F6746 | GCGGTGGATTGTAGACCCATARACAGAGGT |
| Z-Ile | AAGGRHYACTTTGATAGAG | FND5-1180 | ATRATNGCRTCYTTDGARWARAANCC |
| ZND5-1050 | GCNATRCTNTTYYTNTGYTCNGGNTC | F-Cytb | TGNCCRATRAKVAYRWADGGRTSTTC |
| ZND6-210 | GCHARNGCNRCHGARTANGCAAA | FCOI-70 | CCHACYATNCCDGCYCARGCMCCRAA |
| Z6468 | CCACATCTDCTGCATGCAAAYCAYACACTT | FATP6-495 | AGRTGNCCNGCNGTBARRTTNGCNGT |
| ZATP-310 | ACHTTYACNCCHACHACNCARCTNTC | FND4-865 | CCYATGTGVCYNACDGADGAGTADGC |
| Z10818 | TTYGAAGCAGCCGCMTGATACTGACAYTT | F13413 | TAGCTGCTACTCGGATTTGCACCAAGAGT |
| Z13347 | AAGGATAACAGCTCATCCGTTGGTCTTAGG | F49 | GGCCCATCTTAACATCTTC |
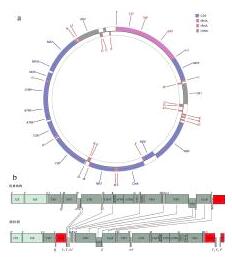
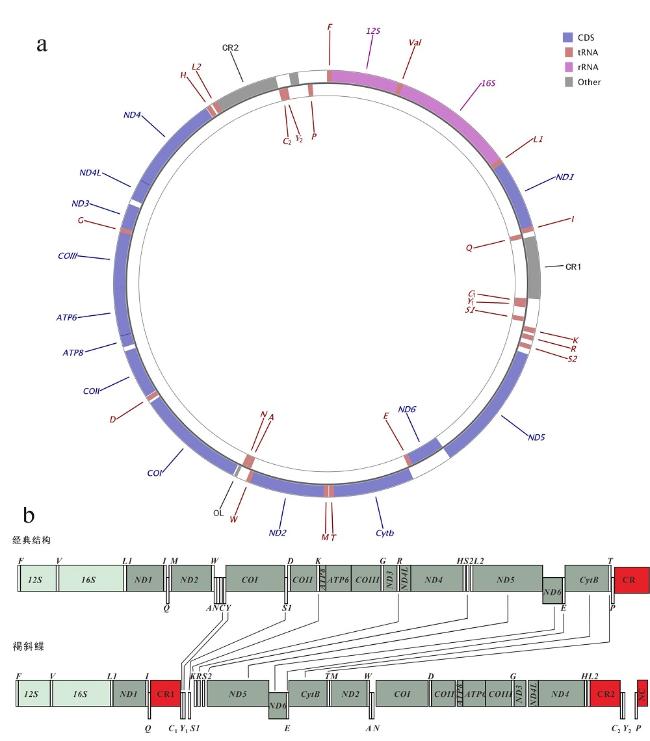
Fig. 1 Gene map of P. glossa mitogenomeand the features of gene rearrangement. (a) Gene map of P. glossa mitogenome; (b) the features of gene rearrangement. repeat region representes repeat sequence. Protein-coding genes are indicated by dark green boxes, rRNA by light green boxes, CRs (control region) and NC (non-coding region) by red boxes, and tRNA by white columns. The genes and their full names are listed in |
Tab. 2 Organization of P. glossa mitochondrial genome表2 褐斜鲽线粒体基因组结构 |
| 基因 | 位置 | 长度 | 密码子 | 间隔区 | |||||
|---|---|---|---|---|---|---|---|---|---|
| 起始 | 终止 | bp | aa | 起始 | 终止 | 反密码子 | 编码链 | ||
| Phe(F) | 1 | 68 | 68 | GAA | H | 0 | |||
| 12S | 69 | 1019 | 951 | H | 0 | ||||
| Val(V) | 1020 | 1091 | 72 | TAC | H | 0 | |||
| 16S | 1092 | 2814 | 1723 | H | 0 | ||||
| Leu1(L1) | 2815 | 2888 | 74 | TAA | H | 0 | |||
| ND1 | 2889 | 3863 | 975 | 324 | ATG | TAA | H | 0 | |
| Ile(I) | 3864 | 3935 | 72 | GAT | H | 0 | |||
| Gln(Q) | 3934 | 4005 | 72 | TTG | L | -2 | |||
| CR1 | 4006 | 4898 | 893 | H | 0 | ||||
| Cys1(C1) | 4899 | 4966 | 68 | GCA | L | 0 | |||
| Tyr1(Y1) | 4967 | 5034 | 68 | GTA | L | 0 | |||
| Ser1(S1) | 5176 | 5246 | 71 | TGA | L | 141 | |||
| Lys(K) | 5309 | 5383 | 75 | TTT | H | 62 | |||
| Arg(R) | 5418 | 5487 | 70 | TCG | H | 34 | |||
| Ser2(S2) | 5549 | 5615 | 67 | GCT | H | 61 | |||
| ND5 | 5691 | 7529 | 1839 | 612 | ATG | TAA | H | 75 | |
| ND6 | 7526 | 8047 | 522 | 173 | ATG | TAG | L | -4 | |
| Glu(E) | 8049 | 8117 | 69 | TTC | L | 1 | |||
| Cytb | 8122 | 9262 | 1141 | 380 | ATG | T | H | 4 | |
| Thr(T) | 9263 | 9333 | 71 | TGT | H | 0 | |||
| Met(M) | 9341 | 9409 | 69 | CAT | H | 7 | |||
| ND2 | 9410 | 10456 | 1047 | 348 | ATG | TAG | H | 0 | |
| Trp(W) | 10455 | 10524 | 70 | TCA | H | -2 | |||
| Ala(A) | 10526 | 10593 | 68 | TGC | L | 1 | |||
| Asn(N) | 10595 | 10667 | 73 | GTT | L | 1 | |||
| OL | 10663 | 10706 | 44 | L | -5 | ||||
| COⅠ | 10724 | 12274 | 1551 | 516 | GTG | TAA | H | 17 | |
| Asp(D) | 12306 | 12373 | 68 | GTC | H | 31 | |||
| COⅡ | 12381 | 13079 | 699 | 232 | ATG | AGA | H | 7 | |
| ATP8 | 13141 | 13308 | 168 | 55 | ATG | TAA | H | 61 | |
| ATP6 | 13299 | 13982 | 684 | 227 | ATG | TAA | H | -10 | |
| COⅢ | 13982 | 14767 | 786 | 261 | ATG | TAA | H | -1 | |
| Gly(G) | 14767 | 14836 | 70 | TCC | H | -1 | |||
| ND3 | 14837 | 15187 | 351 | 116 | ATG | TAA | H | 0 | |
| ND4L | 15261 | 15557 | 297 | 98 | ATG | TAA | H | 73 | |
| ND4 | 15551 | 16924 | 1374 | 457 | ATG | AGA | H | -7 | |
| His(H) | 16932 | 17000 | 69 | GTG | H | 7 | |||
| Leu2(L2) | 17028 | 17100 | 73 | TAG | H | 27 | |||
| CR2 | 17101 | 17997 | 897 | H | 0 | ||||
| Cys2(C2) | 17998 | 18065 | 68 | GCA | L | 0 | |||
| Tyr2(Y2) | 18066 | 18133 | 68 | GTA | L | 0 | |||
| Pro(P) | 18436 | 18506 | 71 | TGG | L | 302 | |||
| NC | 18507 | 18723 | 217 | H | 0 | ||||
注: 基因后括号中的字母代表该基因的缩写; 间隔区中的负值表示该基因与上边基因间的碱基重叠数; NC表示非编码区序列 |
Fig. 2 Aligned sequences of the control regions of P. glossa and seven other flatfish. CR1 and CR2: the two control regions in P. glossa mitogenome; Sla: Samariscus latus, NC_024263; Pol: Paralichthys olivaceus, NC_002386; Pco: Pleuronichthys cornutus NC_022445; Hhi: Hippoglossus hippoglossus, NC_009709; Hst: Hippoglossus stenolepis, NC_009710; Rhi: Reinhardtius hippoglossoides, NC_009711; Vmo: Verasper moseri, NC_008461. The shaded sequences represent the conservative blocks. TAS: terminal associated sequence; CSB: conserved sequence block图2 褐斜鲽控制区和其他7种鲽形目鱼类的控制区序列特征比较 |
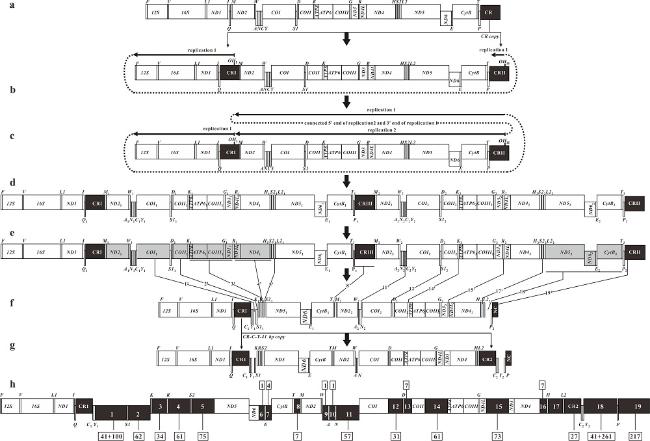
Fig. 3 The inferred mechanism of gene rearrangement from the ancestral gene order to that of P. glossa mitogenome based on the DRRL model. Protein-coding genes and rRNA are indicated by boxes, and tRNA genes are indicated by columns. Genes labeled above the diagram are encoded by the H-strand, and the others, by the L-strand. OH indicates the origin of replication for the H-strand; the direction of replication is shown by arrows. The dark boxes indicate the control regions (CRs), noncoding region (NC) and intergenic spacers. (a) The ancestral mitogenome with 37 genes and one CR. (b) The ancestral CR was duplicated to CRⅠ and CRⅡ, and then CRⅠ was translocated to the position between tRNA-Q and tRNA-M. A first mitochondrial replication event (RP1) was initiated at OHⅠ. (c) After RP1 passed through OHⅡ, secondary replication (RP2) began at OHⅡ. Both replications terminated close to the site of OHⅠ. (d) The duplication of 29 genes and one CR yielded by double replications. (e) One of each copied gene pair was lost randomly. Dark gray boxes indicate degenerated genes. The italic lines indicate the positions of intergenetic spaces from the degenerated genes. The numbers marked by prime indicate the order of the intergenetic spaces in genenome. (f) The order of genes, CRs and intergenic spaces after the loss of several genes. (g) The mitogenome of P. glossa was formed after CRⅠ-tRNA-C1-Y1 and the first 41 bp of Non-coding sequence 1 duplicated and translocated to the position between tRNA-L2 and tRNA-P. (h) The positions and lengths of intergenic spacers of P. glossa. The number in black shaded boxes indicate the intergenic spacer order in mitogenome. The numbers in the boxes above and below represent the length of the intergenic spacers. The boxes above the diagram indicate the sites of intergenic spacers same as the typical one in teleosts; the others are only in P. glossa图3 应用双复制随机丢失模型推测的褐斜鲽线粒体基因重排过程 |
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}