
Journal of Tropical Oceanography >
Genetic Structure and Diversity Analysis of Three Natural Populations of Tectus pyramis Based on Specific Locus Amplified Fragment Sequencing*
|
HUANG Jing (1993—). E-mail: |
Copy editor: YIN Bo
Received date: 2019-12-22
Request revised date: 2020-02-28
Online published: 2020-02-28
Supported by
Strategic Priority Research Program of the Chinese Academy of Sciences(XDA13020206)
Innovation Academy of South China Sea Ecology and Environmental, Engineering, Chinese Academy of Sciences(ISEE2018PY03)
Innovation Academy of South China Sea Ecology and Environmental, Engineering, Chinese Academy of Sciences(ISEE2018ZD02)
Science and Technology Service Network Initiative of Chinese Academy of Sciences(KFJ-STS-ZDTP-055)
Science and Technology Planning Project of Guangdong Province, China(2017B030314052)
Copyright
The marine mollusc Tectus pyramis inhabits the coral reef ecosystems surrounding Dapeng Bay in Shenzhen (SZ), Hainan Island, Xisha (XS) and Nansha Islands of China. In this study, the genetic diversity and genetic structure of three natural populations of T. pyramis from SZ, Sanya (SY) and XS were assessed using specific locus amplified fragment (SLAF) sequencing. A total of 115.74 Mb reads was obtained, with an average depth of 337.87-fold, and an average Q30 value of 94.07 %. Based on 118679 polymorphic SLAF tags, we obtained 846502 highly consistent population single-nucleotide polymorphisms (SNPs), and found 155 SNPs displayed significant differences between populations. Average observed (Ho) and expected (He) heterozygosity values for the three populations ranged from 0.1441 to 0.1611 and from 0.2537 to 0.2695, respectively. Average polymorphism information content (PIC) values were between 0.2048 and 0.2176. The 45 individuals were roughly divided into three groups by phylogenetic tree analysis, principal component analysis (PCA) and clustering; and all the individuals from each population could be clustered into a single group. The genetic structures of SZ and SY populations were similar, yielding the lowest genetic distance (0.2510) and Fst value (0.0818). However, genetic differentiation between XS and the other two populations was significant, especially between SX and SZ (Fst = 0.1868). Genetic differentiation of the three populations may be correlated to ocean currents and geographical locations. The results of this study provide a theoretical basis for germplasm resource management and future aquaculture breeding programs.
HUANG Jing , OU Zhekui , LIU Wenguang , HE Maoxian . Genetic Structure and Diversity Analysis of Three Natural Populations of Tectus pyramis Based on Specific Locus Amplified Fragment Sequencing*[J]. Journal of Tropical Oceanography, 2020 , 39(5) : 1 -18 . DOI: 10.11978/2019136
Tab. 1 Sampling site of Tectus pyramis |
| Population Names | Location | Latitude | Longitude |
|---|---|---|---|
| SZ | Shenzhen | 114°28′23.53″N | 22°31′24.59″E |
| SY | Sanya | 109°26′47.04″N | 18°12′01.76″E |
| XS | Xisha | 112°21′06.09″N | 16°49′10.46″E |
Tab. A1 Sequencing data statistics of the 45 Tectus pyramis |
| Sample ID | Total Reads Number | GC Percentage / % | Q30 Percentage / % |
|---|---|---|---|
| SZ1 | 2646767 | 38.73 | 94.04 |
| SZ2 | 2868617 | 38.82 | 93.27 |
| SZ3 | 3060839 | 38.49 | 93.71 |
| SZ4 | 2928250 | 38.94 | 94.69 |
| SZ5 | 3236397 | 37.86 | 94.48 |
| SZ6 | 3043836 | 38.27 | 94.40 |
| SZ7 | 2817405 | 38.63 | 94.61 |
| SZ8 | 2655334 | 38.56 | 94.77 |
| SZ9 | 2725248 | 38.29 | 94.58 |
| SZ10 | 3277567 | 37.43 | 94.59 |
| SZ11 | 3492934 | 38.09 | 94.81 |
| SZ12 | 3137716 | 38.41 | 94.67 |
| SZ13 | 2039042 | 39.02 | 94.27 |
| SZ14 | 1444547 | 39.31 | 94.19 |
| SZ15 | 1654279 | 39.17 | 93.96 |
| SY1 | 1618225 | 39.35 | 94.28 |
| SY2 | 1683896 | 39.19 | 93.37 |
| SY3 | 1495848 | 39.33 | 93.70 |
| SY4 | 1645635 | 39.30 | 93.83 |
| SY5 | 1832431 | 38.72 | 94.03 |
| SY6 | 2114769 | 38.64 | 94.02 |
| SY7 | 2547407 | 37.89 | 93.08 |
| SY8 | 2000207 | 38.68 | 94.10 |
| SY9 | 2194326 | 38.79 | 94.68 |
| SY10 | 1967775 | 39.10 | 94.86 |
| SY11 | 1841572 | 39.09 | 94.93 |
| SY12 | 2123797 | 38.81 | 94.87 |
| SY13 | 2151154 | 39.17 | 94.85 |
| SY14 | 1954747 | 38.74 | 94.66 |
| SY15 | 2514960 | 37.97 | 93.42 |
| XS1 | 2060640 | 38.78 | 94.46 |
| XS2 | 3164280 | 38.32 | 93.82 |
| XS3 | 3272003 | 38.32 | 94.18 |
| XS4 | 3171569 | 37.92 | 93.92 |
| XS5 | 3202603 | 38.38 | 94.07 |
| XS6 | 2744002 | 38.28 | 93.71 |
| XS7 | 2303163 | 38.29 | 93.63 |
| XS8 | 3218785 | 37.93 | 93.95 |
| XS9 | 2972343 | 38.81 | 94.23 |
| XS10 | 2844445 | 37.76 | 92.61 |
| XS11 | 3272801 | 38.08 | 93.30 |
| XS12 | 3035468 | 38.46 | 93.76 |
| XS13 | 2943254 | 38.05 | 93.17 |
| XS14 | 2583726 | 38.19 | 93.10 |
| XS15 | 3196614 | 37.88 | 93.54 |
| Control | 1046368 | 43.03 | 92.64 |
Note: Total Reads: number of reads for each sample; GC percentage: percentage of total bases of G and C in sequencing results; Q30 percentage: percentage of bases whose sequencing quality value is greater than or equal to 30; Control: data of Oryza sativa ssp. japonica for evaluating experimental database construction; SZ (1 to 15): SZ population; SY (1 to 15): SY population; XS (1 to 15): XS population |
Tab. A2 SLAF tags statistics of the 45 Tectus pyramis |
| Sample ID | SLAF number | Total depth | Average depth |
|---|---|---|---|
| SZ1 | 101537 | 1843808 | 18.16 |
| SZ2 | 101886 | 2006414 | 19.69 |
| SZ3 | 103244 | 2188358 | 21.20 |
| SZ4 | 107505 | 2029284 | 18.88 |
| SZ5 | 108067 | 2338011 | 21.63 |
| SZ6 | 107524 | 2135565 | 19.86 |
| SZ7 | 106819 | 1963328 | 18.38 |
| SZ8 | 109780 | 1860332 | 16.95 |
| SZ9 | 108621 | 1894662 | 17.44 |
| SZ10 | 106877 | 2366741 | 22.14 |
| SZ11 | 113736 | 2502851 | 22.01 |
| SZ12 | 109751 | 2190045 | 19.95 |
| SZ13 | 103761 | 1101996 | 10.62 |
| SZ14 | 98465 | 781192 | 7.93 |
| SZ15 | 100450 | 937431 | 9.33 |
| SY1 | 97381 | 954691 | 9.80 |
| SY2 | 98004 | 1017468 | 10.38 |
| SY3 | 95481 | 909506 | 9.53 |
| SY4 | 96922 | 996596 | 10.28 |
| SY5 | 99774 | 1213282 | 12.16 |
| SY6 | 101941 | 1350287 | 13.25 |
| SY7 | 103004 | 1781398 | 17.29 |
| SY8 | 101915 | 1228982 | 12.06 |
| SY9 | 102675 | 1319548 | 12.85 |
| SY10 | 101263 | 1145948 | 11.32 |
| SY11 | 101126 | 1068060 | 10.56 |
| SY12 | 103524 | 1246384 | 12.04 |
| SY13 | 103145 | 1282700 | 12.44 |
| SY14 | 103038 | 1148245 | 11.14 |
| SY15 | 106571 | 1495169 | 14.03 |
| XS1 | 103543 | 1212705 | 11.71 |
| XS2 | 113977 | 2033850 | 17.84 |
| XS3 | 113800 | 2082711 | 18.30 |
| XS4 | 113523 | 2059873 | 18.14 |
| XS5 | 114052 | 2075178 | 18.20 |
| XS6 | 112858 | 1763198 | 15.62 |
| XS7 | 109594 | 1472432 | 13.44 |
| XS8 | 113536 | 2157654 | 19.00 |
| XS9 | 112609 | 1938036 | 17.21 |
| XS10 | 111522 | 1808792 | 16.22 |
| XS11 | 113827 | 1923615 | 16.90 |
| XS12 | 112561 | 1792407 | 15.92 |
| XS13 | 112568 | 1747609 | 15.52 |
| XS14 | 110288 | 1502944 | 13.63 |
| XS15 | 113093 | 1927904 | 17.05 |
| Total | 218422 | 73797190 | 337.87 |
Note: Sample ID: Sample number; SLAF number: SLAF tag number of corresponding samples; Total depth: total depth of sequencing in SLAF tag of corresponding samples, that is, total reads number; Average depth: average number of sequencing reads of corresponding samples on each SLAF |
Tab. A3 SNP information statistics of the 45 Tectus pyramis |
| Sample ID | Total SNP number | SNP number | Hetloci ratio / % | Integrity ratio / % |
|---|---|---|---|---|
| SZ1 | 2010144 | 1101454 | 10.65 | 54.79 |
| SZ2 | 2010144 | 1108987 | 10.99 | 55.16 |
| SZ3 | 2010144 | 1124233 | 11.03 | 55.92 |
| SZ4 | 2010144 | 1225287 | 11.18 | 60.95 |
| SZ5 | 2010144 | 1199917 | 11.03 | 59.69 |
| SZ6 | 2010144 | 1231716 | 11.21 | 61.27 |
| SZ7 | 2010144 | 1205136 | 10.93 | 59.95 |
| SZ8 | 2010144 | 1185877 | 11.65 | 58.99 |
| SZ9 | 2010144 | 1206178 | 11.23 | 60.00 |
| SZ10 | 2010144 | 1192230 | 10.94 | 59.31 |
| SZ11 | 2010144 | 1283751 | 12.85 | 63.86 |
| SZ12 | 2010144 | 1249074 | 11.64 | 62.13 |
| SZ13 | 2010144 | 1222192 | 10.91 | 60.80 |
| SZ14 | 2010144 | 1061836 | 9.44 | 52.82 |
| SZ15 | 2010144 | 1102628 | 9.75 | 54.85 |
| SY1 | 2010144 | 1038006 | 9.95 | 51.63 |
| SY2 | 2010144 | 1039742 | 9.99 | 51.72 |
| SY3 | 2010144 | 991652 | 9.56 | 49.33 |
| SY4 | 2010144 | 1027875 | 9.87 | 51.13 |
| SY5 | 2010144 | 1040267 | 10.05 | 51.75 |
| SY6 | 2010144 | 1122866 | 10.85 | 55.85 |
| SY7 | 2010144 | 1097668 | 11.22 | 54.60 |
| SY8 | 2010144 | 1126900 | 10.79 | 56.06 |
| SY9 | 2010144 | 1164083 | 11.26 | 57.91 |
| SY10 | 2010144 | 1138116 | 10.73 | 56.61 |
| SY11 | 2010144 | 1130375 | 10.70 | 56.23 |
| SY12 | 2010144 | 1182425 | 11.26 | 58.82 |
| SY13 | 2010144 | 1154786 | 11.52 | 57.44 |
| SY14 | 2010144 | 1155444 | 11.03 | 57.48 |
| SY15 | 2010144 | 1223114 | 12.02 | 60.84 |
| XS1 | 2010144 | 1149640 | 11.35 | 57.19 |
| XS2 | 2010144 | 1300295 | 13.19 | 64.68 |
| XS3 | 2010144 | 1307590 | 13.18 | 65.04 |
| XS4 | 2010144 | 1295832 | 13.14 | 64.46 |
| XS5 | 2010144 | 1310729 | 13.41 | 65.20 |
| XS6 | 2010144 | 1269962 | 12.86 | 63.17 |
| XS7 | 2010144 | 1197594 | 11.65 | 59.57 |
| XS8 | 2010144 | 1268115 | 12.84 | 63.08 |
| XS9 | 2010144 | 1267870 | 12.57 | 63.07 |
| XS10 | 2010144 | 1235461 | 12.47 | 61.46 |
| XS11 | 2010144 | 1313024 | 13.94 | 65.31 |
| XS12 | 2010144 | 1287328 | 13.38 | 64.04 |
| XS13 | 2010144 | 1280442 | 13.48 | 63.69 |
| XS14 | 2010144 | 1253266 | 12.95 | 62.34 |
| XS15 | 2010144 | 1293659 | 13.75 | 64.35 |
Note: Sample ID: Sample number; Total SNP: total SNP detected; SNP num: number of SNP detected in corresponding samples; Integrity: SNP integrity detected in samples; Heter ratio: heterozygosity of SNP detected in samples |
Tab. A4 Significant differences population SNPs Statistics of three populations |
| Marker | Pos | p-value (SY : XS) | p-value (SY : SZ) | p-value (XS : SZ) |
|---|---|---|---|---|
| Marker3275 | 8 | 5.97×10-4 | 3.32×10-5 | 2.09×10-12 |
| Marker5368 | 96 | 6.42×10-5 | 5.24×10-16 | 5.83×10-6 |
| Marker16106 | 153 | 2.51×10-4 | 8.04×10-5 | 8.21×10-13 |
| Marker29414 | 71 | 8.05×10-5 | 8.05×10-5 | 1.52×10-14 |
| Marker31028 | 126 | 1.30×10-4 | 8.24×10-4 | 7.82×10-12 |
| Marker32764 | 118 | 5.54×10-13 | 1.10×10-5 | 9.93×10-4 |
| Marker35459 | 119 | 1.72×10-10 | 3.60×10-4 | 5.97×10-4 |
| Marker39352 | 151 | 3.28×10-5 | 6.11×10-4 | 2.02×10-13 |
| Marker40948 | 162 | 5.83×10-12 | 9.06×10-4 | 3.33×10-4 |
| Marker43655 | 114 | 5.19×10-5 | 9.18×10-6 | 5.24×10-16 |
| Marker43655 | 195 | 2.46×10-4 | 1.89×10-6 | 5.24×10-16 |
| Marker48466 | 76 | 9.12×10-4 | 4.49×10-4 | 4.79×10-11 |
| Marker54309 | 156 | 7.97×10-4 | 6.72×10-4 | 6.70×10-11 |
| Marker56258 | 144 | 9.77×10-4 | 2.29×10-6 | 1.54×10-14 |
| Marker56258 | 153 | 9.77×10-4 | 2.29×10-6 | 1.54×10-14 |
| Marker56258 | 177 | 9.77×10-4 | 2.29×10-6 | 1.54×10-14 |
| Marker56258 | 187 | 9.77×10-4 | 2.29×10-6 | 1.54×10-14 |
| Marker56258 | 196 | 9.77×10-4 | 2.29×10-6 | 1.54×10-14 |
| Marker57704 | 38 | 1.83×10-12 | 8.31×10-5 | 4.12×10-4 |
| Marker57704 | 56 | 1.83×10-12 | 8.31×10-5 | 4.12×10-4 |
| Marker58004 | 63 | 7.97×10-4 | 1.10×10-5 | 9.23×10-14 |
| Marker58004 | 192 | 7.97×10-4 | 1.10×10-5 | 9.23×10-14 |
| Marker58561 | 4 | 1.78×10-14 | 2.59×10-6 | 4.78×10-4 |
| Marker58831 | 165 | 7.08×10-4 | 9.92×10-5 | 1.01×10-11 |
| Marker58831 | 179 | 7.08×10-4 | 9.92×10-5 | 1.01×10-11 |
| Marker59195 | 168 | 8.50×10-14 | 3.92×10-5 | 5.09×10-4 |
| Marker61327 | 50 | 4.25×10-4 | 5.13×10-4 | 1.01×10-12 |
| Marker62076 | 121 | 9.36×10-5 | 3.65×10-5 | 2.88×10-14 |
| Marker62305 | 23 | 4.53×10-5 | 7.45×10-5 | 8.23×10-15 |
| Marker64435 | 95 | 5.78×10-5 | 3.54×10-13 | 7.07×10-4 |
| Marker64938 | 164 | 1.26×10-6 | 1.29×10-4 | 5.24×10-16 |
| Marker64938 | 192 | 1.26×10-6 | 1.29×10-4 | 5.24×10-16 |
| Marker65298 | 19 | 1.26×10-6 | 4.05×10-4 | 4.66×10-15 |
| Marker72476 | 169 | 7.75×10-5 | 9.41×10-4 | 4.08×10-11 |
| Marker74137 | 39 | 9.84×10-4 | 4.89×10-6 | 1.78×10-14 |
| Marker74137 | 160 | 4.09×10-5 | 1.66×10-5 | 7.13×10-16 |
| Marker74137 | 197 | 4.09×10-5 | 1.66×10-5 | 7.13×10-16 |
| Marker77452 | 27 | 3.34×10-4 | 1.62×10-4 | 9.40×10-12 |
| Marker77571 | 174 | 7.37×10-4 | 4.81×10-7 | 2.00×10-15 |
| Marker78614 | 80 | 9.63×10-4 | 9.43×10-5 | 5.49×10-12 |
| Marker79010 | 46 | 5.56×10-6 | 5.96×10-4 | 1.13×10-14 |
| Marker79010 | 96 | 5.56×10-6 | 5.96×10-4 | 1.13×10-14 |
| Marker80829 | 28 | 4.18×10-5 | 6.72×10-4 | 9.86×10-13 |
| Marker85121 | 127 | 2.97×10-4 | 1.04×10-4 | 2.02×10-13 |
| Marker86368 | 91 | 3.80×10-4 | 1.03×10-9 | 9.11×10-4 |
| Marker89723 | 73 | 2.82×10-5 | 4.83×10-4 | 3.55×10-13 |
| Marker89828 | 32 | 7.08×10-4 | 8.14×10-4 | 2.12×10-9 |
| Marker91169 | 142 | 9.10×10-4 | 1.61×10-4 | 9.61×10-12 |
| Marker91169 | 143 | 9.10×10-4 | 3.97×10-4 | 2.55×10-11 |
| Marker91169 | 193 | 9.10×10-4 | 1.61×10-4 | 9.61×10-12 |
| Marker93033 | 35 | 5.09×10-4 | 3.39×10-4 | 5.69×10-12 |
| Marker94025 | 137 | 8.55×10-4 | 4.15×10-4 | 3.54×10-13 |
| Marker96746 | 95 | 3.15×10-4 | 6.99×10-4 | 5.37×10-11 |
| Marker97131 | 48 | 9.89×10-5 | 9.28×10-6 | 4.03×10-15 |
| Marker97131 | 64 | 9.89×10-5 | 1.66×10-5 | 8.23×10-15 |
| Marker97651 | 49 | 3.07×10-4 | 2.13×10-4 | 1.38×10-11 |
| Marker99962 | 152 | 6.07×10-5 | 4.60×10-4 | 2.87×10-12 |
| Marker101446 | 75 | 2.31×10-4 | 4.98×10-4 | 6.74×10-11 |
| Marker102130 | 123 | 2.82×10-5 | 6.46×10-4 | 2.32×10-13 |
| Marker103561 | 24 | 5.74×10-4 | 7.97×10-4 | 3.29×10-11 |
| Marker106244 | 7 | 3.73×10-4 | 1.53×10-4 | 1.18×10-13 |
| Marker114322 | 156 | 2.85×10-5 | 8.53×10-4 | 2.44×10-12 |
| Marker114487 | 160 | 9.77×10-4 | 2.06×10-4 | 6.96×10-11 |
| Marker115710 | 9 | 3.72×10-4 | 7.06×10-5 | 4.07×10-13 |
| Marker115710 | 37 | 3.72×10-4 | 4.05×10-4 | 9.61×10-12 |
| Marker116654 | 73 | 1.91×10-4 | 8.85×10-5 | 2.22×10-13 |
| Marker120806 | 120 | 3.64×10-4 | 7.18×10-10 | 4.58×10-4 |
| Marker128452 | 23 | 4.11×10-13 | 2.60×10-4 | 8.79×10-6 |
| Marker130399 | 64 | 2.87×10-12 | 9.76×10-4 | 2.59×10-5 |
| Marker131746 | 129 | 4.78×10-4 | 6.66×10-4 | 9.09×10-11 |
| Marker134085 | 78 | 1.05×10-11 | 4.50×10-4 | 2.22×10-4 |
| Marker135504 | 57 | 2.29×10-7 | 7.97×10-4 | 5.24×10-16 |
| Marker136107 | 156 | 8.50×10-14 | 6.40×10-4 | 6.95×10-6 |
| Marker142198 | 82 | 4.78×10-4 | 3.68×10-4 | 1.48×10-11 |
| Marker146079 | 23 | 7.48×10-4 | 8.24×10-5 | 1.44×10-12 |
| Marker146079 | 58 | 7.48×10-4 | 8.24×10-5 | 1.44×10-12 |
| Marker149477 | 131 | 8.47×10-4 | 5.31×10-4 | 1.62×10-10 |
| Marker153118 | 160 | 7.93×10-4 | 8.65×10-4 | 6.32×10-10 |
| Marker157365 | 21 | 9.10×10-4 | 1.45×10-4 | 1.62×10-12 |
| Marker164040 | 135 | 7.36×10-4 | 5.21×10-4 | 2.70×10-10 |
| Marker165818 | 206 | 2.35×10-15 | 7.26×10-5 | 1.67×10-4 |
| Marker169492 | 68 | 3.34×10-4 | 1.39×10-4 | 1.25×10-12 |
| Marker170499 | 42 | 2.35×10-4 | 3.12×10-4 | 9.23×10-14 |
| Marker170499 | 61 | 2.35×10-4 | 3.12×10-4 | 9.23×10-14 |
| Marker170499 | 70 | 2.35×10-4 | 3.12×10-4 | 9.23×10-14 |
| Marker170499 | 85 | 2.35×10-4 | 3.12×10-4 | 9.23×10-14 |
| Marker173073 | 161 | 5.88×10-4 | 3.19×10-8 | 3.44×10-17 |
| Marker174886 | 165 | 8.39×10-15 | 3.44×10-6 | 4.12×10-4 |
| Marker174886 | 174 | 8.39×10-15 | 3.44×10-6 | 4.12×10-4 |
| Marker176168 | 67 | 5.49×10-12 | 2.51×10-4 | 3.74×10-4 |
| Marker177413 | 57 | 6.14×10-4 | 1.15×10-4 | 3.74×10-12 |
| Marker181332 | 62 | 1.75×10-4 | 6.40×10-4 | 2.02×10-11 |
| Marker181637 | 22 | 1.98×10-4 | 2.38×10-5 | 2.83×10-12 |
| Marker181637 | 23 | 1.98×10-4 | 2.38×10-5 | 2.83×10-12 |
| Marker181637 | 33 | 4.98×10-4 | 1.24×10-5 | 2.83×10-12 |
| Marker181637 | 36 | 1.98×10-4 | 2.38×10-5 | 2.83×10-12 |
| Marker182931 | 24 | 4.13×10-5 | 2.31×10-4 | 5.69×10-14 |
| Marker182931 | 36 | 4.13×10-5 | 2.31×10-4 | 5.69×10-14 |
| Marker182931 | 38 | 4.99×10-4 | 2.31×10-4 | 1.44×10-12 |
| Marker182931 | 114 | 4.99×10-4 | 4.90×10-4 | 7.37×10-12 |
| Marker182931 | 171 | 4.99×10-4 | 2.31×10-4 | 1.44×10-12 |
| Marker184767 | 188 | 1.95×10-12 | 3.64×10-4 | 2.07×10-5 |
| Marker185952 | 36 | 3.34×10-4 | 2.43×10-4 | 1.89×10-11 |
| Marker186713 | 35 | 1.46×10-6 | 3.41×10-5 | 3.44×10-17 |
| Marker190981 | 52 | 4.12×10-4 | 8.23×10-5 | 7.84×10-13 |
| Marker192459 | 199 | 7.33×10-4 | 3.39×10-4 | 1.88×10-11 |
| Marker195736 | 85 | 1.72×10-4 | 7.11×10-4 | 1.59×10-11 |
| Marker195973 | 95 | 7.07×10-4 | 5.24×10-16 | 4.75×10-7 |
| Marker197784 | 195 | 8.47×10-4 | 5.80×10-4 | 3.01×10-11 |
| Marker202019 | 15 | 5.21×10-4 | 1.19×10-4 | 8.82×10-12 |
| Marker202019 | 84 | 5.21×10-4 | 1.19×10-4 | 8.82×10-12 |
| Marker203633 | 31 | 2.19×10-11 | 7.35×10-4 | 5.69×10-4 |
| Marker204262 | 11 | 4.11×10-5 | 7.37×10-4 | 1.89×10-11 |
| Marker204262 | 76 | 6.80×10-4 | 7.37×10-4 | 5.49×10-10 |
| Marker204262 | 145 | 4.11×10-5 | 7.37×10-4 | 1.89×10-11 |
| Marker204262 | 159 | 4.11×10-5 | 7.37×10-4 | 1.89×10-11 |
| Marker204262 | 162 | 4.11×10-5 | 7.37×10-4 | 1.89×10-11 |
| Marker204262 | 169 | 4.11×10-5 | 7.37×10-4 | 1.89×10-11 |
| Marker204894 | 204 | 4.99×10-4 | 4.90×10-4 | 7.37×10-12 |
| Marker215169 | 49 | 2.59×10-5 | 9.10×10-4 | 8.12×10-13 |
| Marker217857 | 37 | 7.82×10-4 | 5.13×10-4 | 6.01×10-12 |
| Marker217857 | 81 | 7.82×10-4 | 5.13×10-4 | 6.01×10-12 |
| Marker219928 | 157 | 1.15×10-13 | 6.49×10-4 | 5.18×10-6 |
| Marker220971 | 29 | 8.47×10-4 | 2.38×10-4 | 7.37×10-12 |
| Marker227957 | 59 | 3.55×10-4 | 2.89×10-6 | 1.13×10-14 |
| Marker240685 | 145 | 3.25×10-4 | 1.52×10-4 | 2.22×10-13 |
| Marker253092 | 204 | 9.93×10-4 | 1.10×10-5 | 5.54×10-13 |
| Marker253901 | 155 | 5.53×10-4 | 3.77×10-4 | 4.50×10-11 |
| Marker268253 | 137 | 1.07×10-5 | 3.17×10-12 | 5.97×10-4 |
| Marker273125 | 41 | 8.30×10-4 | 6.05×10-4 | 3.69×10-10 |
| Marker273125 | 51 | 8.30×10-4 | 6.05×10-4 | 3.69×10-10 |
| Marker273125 | 59 | 8.30×10-4 | 6.05×10-4 | 3.69×10-10 |
| Marker273125 | 67 | 8.30×10-4 | 6.05×10-4 | 3.69×10-10 |
| Marker273125 | 147 | 8.30×10-4 | 6.05×10-4 | 3.69×10-10 |
| Marker273125 | 187 | 8.30×10-4 | 6.05×10-4 | 3.69×10-10 |
| Marker280356 | 52 | 8.65×10-4 | 8.85×10-5 | 1.19×10-11 |
| Marker281848 | 160 | 2.56×10-4 | 6.58×10-4 | 1.38×10-11 |
| Marker289553 | 74 | 9.36×10-5 | 2.07×10-5 | 3.65×10-14 |
| Marker289553 | 128 | 9.36×10-5 | 2.07×10-5 | 3.65×10-14 |
| Marker296021 | 144 | 4.18×10-5 | 2.99×10-4 | 2.35×10-15 |
| Marker298184 | 70 | 3.93×10-6 | 2.61×10-5 | 3.44×10-17 |
| Marker308966 | 59 | 8.21×10-13 | 2.49×10-4 | 1.24×10-4 |
| Marker330769 | 199 | 1.58×10-4 | 3.40×10-4 | 2.02×10-11 |
| Marker350607 | 45 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 49 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 71 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 73 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 74 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 82 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 141 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 163 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker350607 | 181 | 2.63×10-10 | 1.48×10-4 | 4.58×10-4 |
| Marker362138 | 51 | 1.48×10-4 | 1.38×10-4 | 8.82×10-12 |
| Marker389768 | 132 | 3.05×10-4 | 7.36×10-4 | 6.74×10-11 |
| Marker475043 | 205 | 1.26×10-5 | 2.37×10-4 | 8.23×10-15 |
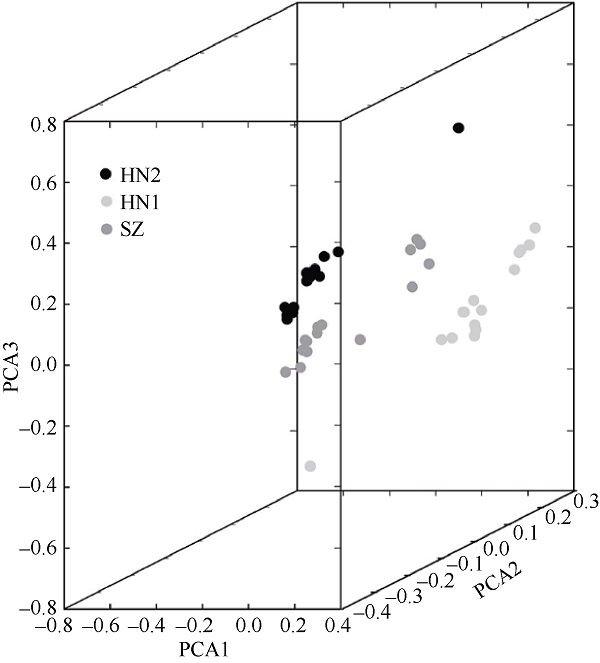
Fig. 3 PCA cluster analysis of 45 Tectus pyramis individuals. Samples are clustered into three dimensions by PCA analysis. PCA1 ~ PCA3 represents the first, second, and third principal components, respectively |
Tab. A5 PCA coefficients of individuals |
| Sample ID | PCA1 | PCA2 | PCA3 |
|---|---|---|---|
| SZ1 | 0.28 | -0.09 | 0.24 |
| SZ2 | 0.29 | -0.08 | 0.22 |
| SZ3 | 0.27 | -0.10 | 0.21 |
| SZ4 | 0.10 | -0.29 | 0.02 |
| SZ5 | 0.05 | -0.28 | -0.07 |
| SZ6 | 0.11 | -0.30 | 0.03 |
| SZ7 | 0.13 | -0.27 | 0.06 |
| SZ8 | 0.15 | -0.27 | 0.07 |
| SZ9 | 0.12 | -0.28 | 0.04 |
| SZ10 | 0.09 | -0.29 | -0.02 |
| SZ11 | 0.06 | -0.33 | -0.06 |
| SZ12 | 0.10 | -0.30 | -0.01 |
| SZ13 | 0.11 | -0.14 | -0.06 |
| SZ14 | 0.20 | 0.00 | 0.11 |
| SZ15 | 0.19 | -0.04 | 0.06 |
| SY1 | 0.26 | 0.24 | 0.04 |
| SY2 | 0.26 | 0.23 | 0.02 |
| SY3 | 0.28 | 0.27 | 0.08 |
| SY4 | 0.28 | 0.25 | 0.05 |
| SY5 | 0.26 | 0.22 | -0.03 |
| SY6 | 0.21 | 0.15 | -0.13 |
| SY7 | 0.19 | 0.16 | -0.16 |
| SY8 | 0.20 | 0.14 | -0.17 |
| SY9 | 0.22 | 0.10 | -0.10 |
| SY10 | 0.22 | 0.13 | -0.08 |
| SY11 | 0.21 | 0.14 | -0.13 |
| SY12 | 0.19 | 0.08 | -0.18 |
| SY13 | 0.21 | 0.11 | -0.11 |
| SY14 | 0.18 | 0.12 | -0.20 |
| SY15 | -0.17 | -0.10 | -0.50 |
| XS1 | -0.03 | 0.25 | 0.41 |
| XS2 | -0.36 | 0.03 | 0.07 |
| XS3 | -0.36 | 0.03 | 0.07 |
| XS4 | -0.36 | 0.04 | 0.05 |
| XS5 | -0.36 | 0.03 | 0.04 |
| XS6 | -0.35 | 0.05 | 0.07 |
| XS7 | -0.33 | 0.10 | 0.10 |
| XS8 | -0.34 | 0.05 | 0.08 |
| XS9 | -0.33 | 0.06 | 0.10 |
| XS10 | -0.36 | 0.07 | 0.03 |
| XS11 | -0.42 | 0.01 | -0.05 |
| XS12 | -0.41 | 0.02 | -0.04 |
| XS13 | -0.42 | 0.02 | -0.05 |
| XS14 | -0.41 | 0.04 | -0.05 |
| XS15 | -0.43 | 0.02 | -0.07 |
Note: Sample ID: Sample number; PCA1: the first principal component; PCA2: the second principal component; PCA3: the third principal component |
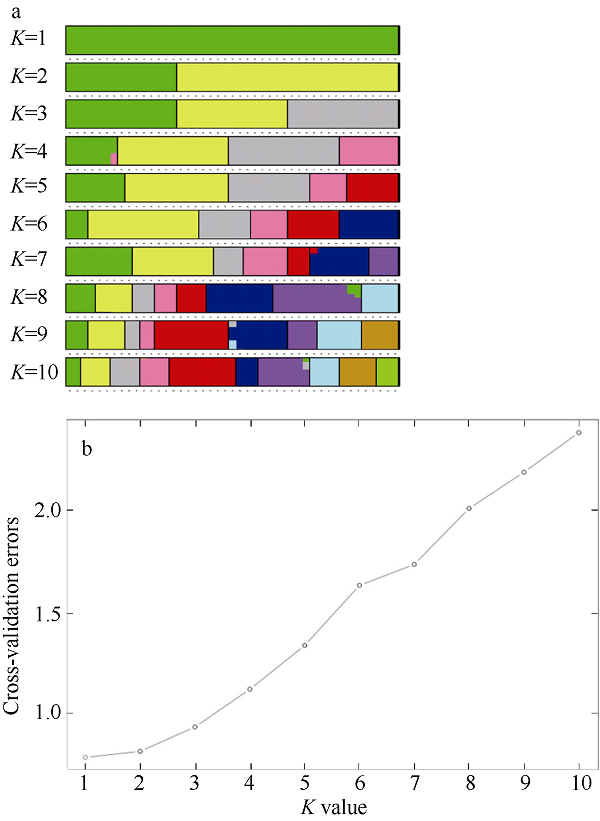
Fig. 4 Admixture individual cluster values corresponding to each K value (a), and admixture validation error rate corresponding to different K values (b). Different color in each line represents different clusters in |
Tab. 2 Genetic diversity of Tectus pyramis among the three populations |
| Parameter | SZ population | SY population | XS population |
|---|---|---|---|
| Na | 1.7719 | 1.8038 | 1.8095 |
| Ne | 1.4254 | 1.4463 | 1.4511 |
| I | 0.3847 | 0.4040 | 0.4081 |
| Ho | 0.1545 | 0.1441 | 0.1611 |
| He | 0.2537 | 0.2666 | 0.2695 |
| PIC | 0.2048 | 0.2152 | 0.2176 |
| MAF | 0.2414 | 0.2431 | 0.2437 |
Note: The average value of genetic diversity for T. pyramis is given. Na, number of observed alleles; Ne, effective number of alleles; I, Shannon information index; Ho, observed heterozygosity; He, expected heterozygosity; PIC, polymorphism information content; MAF, minor allele frequency |
Tab. 3 Genetic structural parameters for the three populations of Tectus pyramis |
| Population | Nm and Fst | D and S | |||||
|---|---|---|---|---|---|---|---|
| SY | XS | SZ | SY | XS | SZ | ||
| SY | — | 1.3914 | 2.8070 | — | 0.7240 | 0.7780 | |
| XS | 0.1523 | — | 1.0882 | 0.3230 | — | 0.7189 | |
| SZ | 0.0818 | 0.1868 | — | 0.2510 | 0.3300 | — | |
Note: For Nm and Fst, data above the diagonal are pairwise Nm values, and data below the diagonal are Fst values. Nm (gene flow, estimated from Fst) = 0.25 (1 - Fst) / Fst. Fst, reflecting inbreeding among subpopulations relative to the total population. For D and S, data above the diagonal are the genetic similarity coefficient (S), and data below the diagonal are genetic distance (D). “—” indicates comparison within the same population |
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}